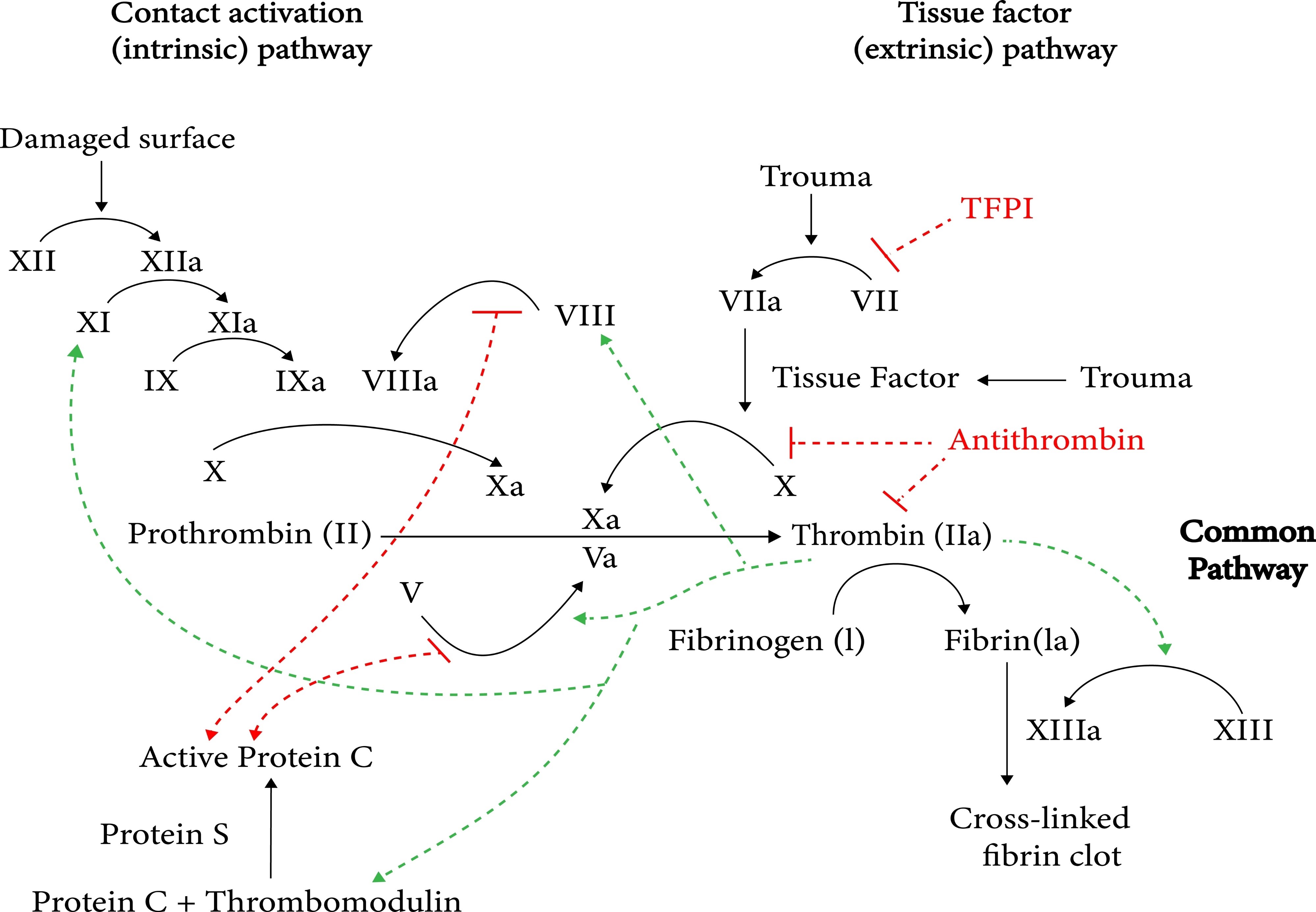
Viruses are obligate intracellular parasites - they can’t replicate on their own and must commandeer host cell machinery. Understanding viral biology helps you understand disease patterns and treatment options.
This chapter organizes viruses by genome type (DNA vs RNA) and family, covers the diagnostic workup from cytopathic effect through molecular methods, and closes with prions and vaccines. For board purposes, keep a mental map of four things per virus: genome (ds/ss, DNA/RNA, +/- sense), envelope status, the classic clinical syndrome, and the preferred test.
A virion is nucleic acid plus a protein capsid, with or without a lipid envelope derived from host membrane. The capsid is built from subunits called capsomeres and comes in three shapes: icosahedral (20 triangular faces, 12 vertices - adenovirus, parvovirus, picornavirus), helical (capsomeres spiral around nucleic acid - influenza, rabies, Ebola), and complex (pox bricks, bacteriophage head-tail).
Genome shape can be linear (most common) or circular (HPV, HBV, polyomavirus). Segmented genomes (influenza, rotavirus, bunyavirus) consist of multiple separate RNA segments, enabling reassortment.
49.1 DNA Viruses
Herpesviruses: Latency Defines the Family
The herpesviruses share a defining characteristic: after primary infection, they establish lifelong latent infection. The virus persists in specific cell types, reactivating periodically to cause recurrent disease. There are eight human herpesviruses, and understanding where each hides and what it does when it awakens is the key to mastering this family.
Structure: all herpesviruses are large, enveloped, double-stranded DNA viruses with an icosahedral nucleocapsid. During latency the viral genome persists as a circular episome in the nucleus with minimal gene expression. Reactivation follows immunosuppression, stress, UV exposure, or fever.
Subfamilies and latency sites:
- Alphaherpesvirinae (HSV-1, HSV-2, VZV): latent in sensory neurons (dorsal root / trigeminal ganglia)
- Betaherpesvirinae (CMV, HHV-6, HHV-7): latent in myeloid/lymphoid cells (monocytes, CD4 T cells)
- Gammaherpesvirinae (EBV, HHV-8): latent in lymphocytes (B cells mostly; HHV-8 also in endothelial cells)
A quick reference table for latency:
| HSV-1 |
Trigeminal ganglion |
Oral herpes, encephalitis (temporal lobe) |
| HSV-2 |
Lumbosacral ganglia (S2-S5) |
Genital herpes, Mollaret meningitis |
| VZV |
Dorsal root ganglia |
Chickenpox, shingles |
| EBV |
B cells (via CD21) |
Mono, Burkitt, NPC, PTLD |
| CMV |
Monocytes, myeloid progenitors |
Mono-like, transplant disease, congenital |
| HHV-6 |
CD4+ T cells |
Roseola |
| HHV-7 |
CD4+ T cells |
Roseola (less common) |
| HHV-8 |
B cells, endothelial cells |
Kaposi sarcoma, PEL, MCD |
HHV-6 can integrate into host chromosomes (telomeric integration) and be inherited vertically. About 1% of the population has chromosomally integrated HHV-6 (ciHHV-6), which causes persistently elevated blood PCR that can be misread as active infection - distinguish with hair follicle PCR.
HSV-1 and HSV-2 both cause vesicular lesions, but HSV-1 classically affects the orolabial area while HSV-2 classically affects the genital area (though either can infect either site). After primary infection, the viruses travel up sensory nerves and establish latency in ganglia - trigeminal ganglia for oral infection, sacral ganglia for genital infection. Reactivation produces the recurrent “cold sores” or genital outbreaks patients experience for life. HSV-1 also causes herpes keratitis (corneal infection that can lead to blindness) and is the most common cause of sporadic encephalitis in adults - a devastating infection that is treatable if recognized and treated promptly with IV acyclovir.
CNS syndromes are a high-yield distinction. HSV-1 causes encephalitis (temporal lobe predilection, the most common sporadic viral encephalitis in adults). HSV-2 causes meningitis, including Mollaret meningitis - a recurrent benign lymphocytic meningitis with episodes lasting 2-5 days separated by symptom-free intervals. Memory hook: HSV-1 = ONE brain (encephalitis, cerebral); HSV-2 = TWO meninges.
HSV-1 encephalitis classically involves bilateral necrotizing lesions of the anterior temporal lobes (and inferior frontal / insular cortex), thought to reflect spread from trigeminal ganglion along tentorial branches or from olfactory pathways. MRI shows T2/FLAIR hyperintensity with hemorrhagic necrosis. Presentation: fever, headache, confusion, aphasia, seizures, behavioral changes. Mortality without treatment exceeds 70%.
Other HSV clinical syndromes:
- Herpetic whitlow - painful vesicular lesion on finger/hand, historically in dentists and respiratory therapists. Do not incise and drain - I&D is contraindicated because it spreads infection.
- Erythema multiforme - HSV (usually HSV-1) and Mycoplasma pneumoniae are the two classic infectious triggers. EM appears 1-3 weeks after a herpes outbreak. Recurrent EM is almost always HSV-associated; prophylactic acyclovir prevents it.
- Neonatal HSV - acquired at vaginal delivery from active maternal genital lesions. Three patterns: SEM (skin/eye/mouth), CNS, disseminated. Disseminated disease carries ~30% mortality even with treatment. Cesarean delivery is indicated if active genital lesions are present at delivery. HSV-2 is the most common cause, but neonatal HSV-1 is increasing.
HSV diagnostics:
- CSF PCR is the gold standard for HSV encephalitis (>95% sensitivity, ~99% specificity). Can be negative very early (within 72 hours) - if suspicion is high, repeat and treat empirically with IV acyclovir.
- Viral culture: HSV grows quickly, within 1-5 days, on MRC5 (human fetal lung fibroblasts), Vero, A549, or rabbit kidney cells. Replaced by PCR for diagnosis but still used for susceptibility testing.
- Tzanck smear - scrape vesicle base, stain with Wright/Giemsa, look for multinucleated giant cells with the Three M’s: Moulding, Margination, Multinucleation. Cannot distinguish HSV from VZV. Sensitivity 60-70%.
- Serology: not useful for diagnosis of acute lesions because seroprevalence is high (50-80% for HSV-1, 15-25% for HSV-2). Type-specific IgG (glycoprotein G-based) can distinguish HSV-1 from HSV-2 for counseling.
Treatment and resistance: Acyclovir and its prodrug valacyclovir are first-line. Acyclovir is a guanosine analog that must be activated by viral thymidine kinase (TK) and then inhibits viral DNA polymerase. Penciclovir/famciclovir work similarly. TK mutations are the most common mechanism of acyclovir resistance. For TK-resistant HSV/VZV, use foscarnet (pyrophosphate analog, directly inhibits DNA polymerase, TK-independent) or cidofovir.
Varicella-zoster virus (VZV) causes chickenpox (varicella) in primary infection - a generalized vesicular rash that appears in crops. The virus then establishes latency in dorsal root ganglia throughout the spine. Decades later, reactivation produces shingles (zoster) - a painful vesicular rash in a dermatomal distribution, limited to the distribution of one sensory nerve. Post-herpetic neuralgia - chronic pain persisting after the rash heals - is a common and debilitating complication.
Chickenpox distribution and stages: rash begins on face/scalp and spreads centrifugally to trunk and extremities, sparing palms and soles. The hallmark is lesions in different stages simultaneously (“crops”) - macules, papules, vesicles, pustules, and crusts all coexisting. The individual vesicle is classically described as a dewdrop on a rose petal. This asynchronous, trunk-predominant pattern distinguishes varicella from smallpox (synchronous, extremity/face predominant).
Zoster reactivation syndromes:
- Classic zoster: unilateral dermatomal vesicles, most often thoracic (T3-L3). Prodromal burning/tingling precedes the rash.
- Postherpetic neuralgia: neuropathic pain persisting >90 days after rash onset. Affects 10-20% of zoster patients, >50% in those over 60. Treatments: gabapentin, pregabalin, TCAs, lidocaine patches. The Shingrix vaccine (recombinant glycoprotein E + AS01B adjuvant) is >90% efficacious and recommended for adults >50.
- Herpes zoster ophthalmicus - V1 distribution, sight-threatening.
- Ramsay Hunt syndrome - VZV reactivation in the geniculate ganglion of CN VII. Triad: ipsilateral facial palsy, vesicles in the ear/canal/TM, and otalgia. Plus hearing loss, tinnitus, vertigo, dysgeusia. Worse prognosis than idiopathic Bell palsy; treat with acyclovir + corticosteroids.
- Disseminated zoster - in immunocompromised, >2 dermatomes or visceral involvement.
VZV diagnostics:
- PCR is the best test - vesicle fluid (swab the base of a vesicle), CSF for CNS disease, BAL for pneumonia, blood for disseminated disease.
- VZV grows slowly in culture (1-4 weeks) on MRC5 fibroblasts - in contrast to HSV’s 1-5 days. VZV is cell-associated (doesn’t release much free virus), so specimens must be inoculated quickly; frozen specimens are suboptimal.
- DFA on vesicle fluid is rapid but less sensitive than PCR.
- IgG serology is useful for confirming immunity (healthcare workers, pregnant women with exposure); IgM is unreliable due to cross-reactivity with HSV.
- Histology shows the same Three M’s (moulding, margination, multinucleation) and Cowdry type A inclusions as HSV - clinical context distinguishes them.
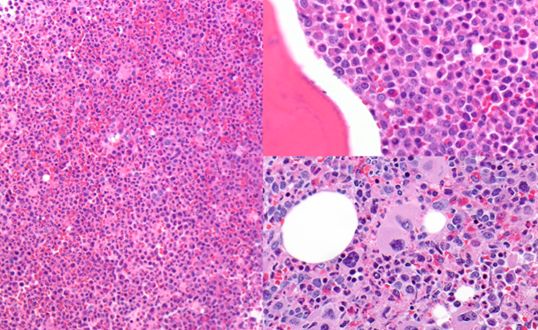
Epstein-Barr virus (EBV) infects B lymphocytes and causes infectious mononucleosis - fever, pharyngitis, lymphadenopathy, and the reactive T cells called “atypical lymphocytes.” EBV establishes latency in B cells. In immunocompromised patients, EBV-driven B cell proliferation can cause post-transplant lymphoproliferative disorder (PTLD) or lymphoma. EBV is also associated with nasopharyngeal carcinoma and Burkitt lymphoma.
Transmission and entry: EBV spreads mostly via saliva (the “kissing disease”), infecting oropharyngeal epithelium first, then B cells. It enters B cells via CD21 (complement receptor 2, CR2), which binds the viral gp350/220 glycoprotein. Shedding in saliva persists for months and then intermittently for life.
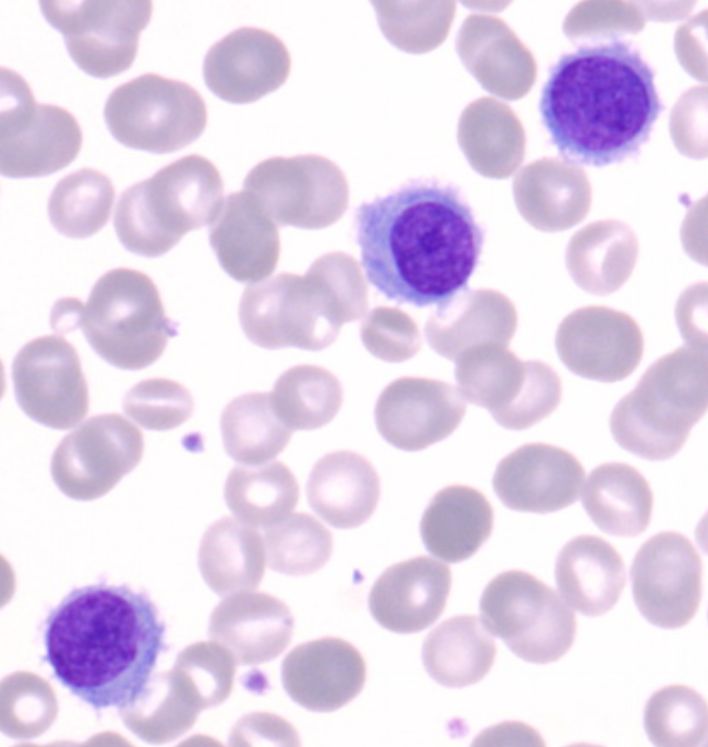
Infectious mononucleosis clinical features: fever, exudative pharyngitis, posterior cervical lymphadenopathy (posterior > anterior - a clue against bacterial pharyngitis), fatigue, hepatosplenomegaly. Spleen rupture is a feared complication - avoid contact sports for 3-4 weeks. A maculopapular rash after ampicillin/amoxicillin occurs in ~90% of mono patients given aminopenicillins and is a classic board association. Labs: lymphocytosis with >10% atypical lymphocytes, elevated transaminases.
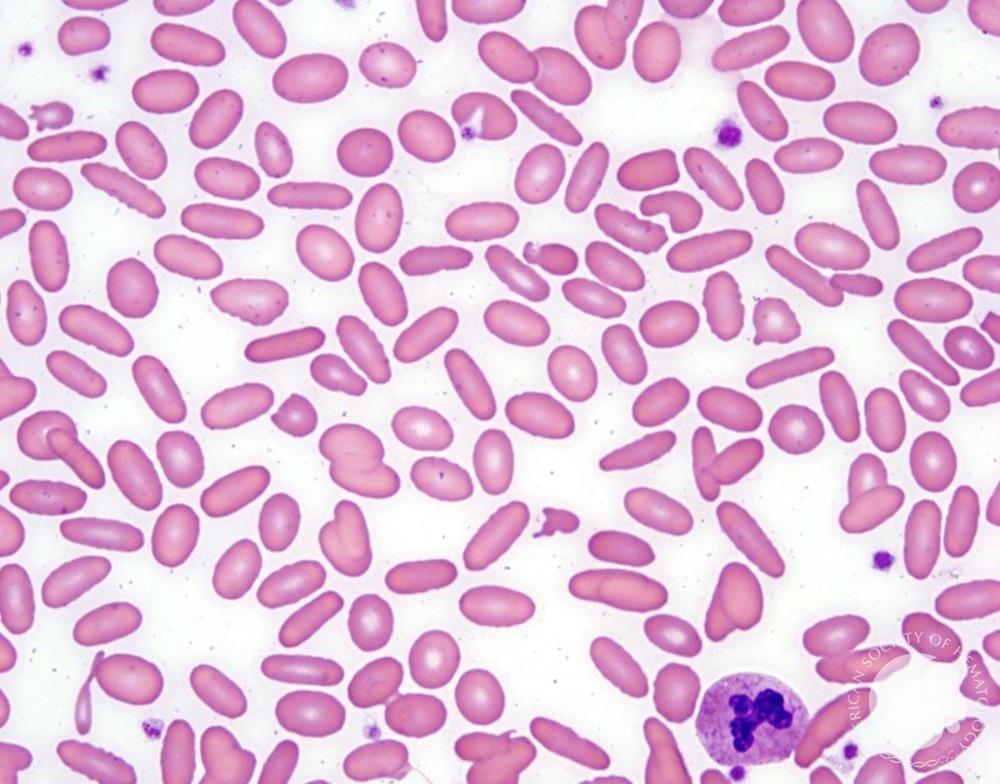
Atypical lymphocytes (Downey type II cells) are reactive CD8+ T cells, not infected B cells. Large, abundant basophilic cytoplasm that scallops around adjacent RBCs. Also seen with CMV, toxoplasmosis, acute HIV, and drug reactions - most prominent with EBV.
Cold agglutinin association: EBV produces IgM anti-i (lowercase, fetal/cord antigen). Memory trick: “i” for Infectious mononucleosis. Contrast with Mycoplasma pneumoniae, which produces anti-I (uppercase) - “I have an atypical pneumonia.”
EBV-associated malignancies (reactivation, especially in immunocompromised):
- PTLD (post-transplant lymphoproliferative disorder) - most common EBV-associated malignancy in transplant
- Burkitt lymphoma (endemic/African form nearly 100% EBV+; sporadic less so) - jaw mass, t(8;14) c-MYC
- Hodgkin lymphoma (mixed-cellularity subtype ~70% EBV+)
- Nasopharyngeal carcinoma (southern China, Southeast Asia)
- NK/T-cell lymphoma, nasal type (nearly 100% EBV+)
- Primary CNS lymphoma in AIDS
- Smooth muscle tumors in immunosuppressed children
- Oral hairy leukoplakia (tongue, AIDS)
X-linked lymphoproliferative disease (Duncan disease): SH2D1A / SAP gene mutation causing fulminant primary EBV infection in young boys, with HLH, B-cell lymphoma, and dysgammaglobulinemia. SAP is needed for normal NK/T-cell cytotoxicity against EBV-infected B cells. Treatment: HSCT.
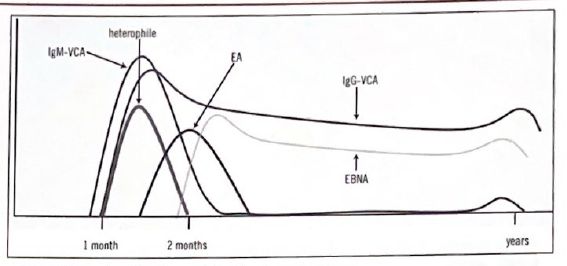
Heterophile antibody test (Monospot)
The Monospot detects heterophile IgM antibodies produced during EBV infection that cross-react with sheep/ox/horse RBC antigens. Modern method is a Davidsohn differential absorption: patient serum is split and absorbed with guinea pig kidney (removes Forssman antibodies but not EBV heterophiles) or beef RBC stroma (removes EBV heterophiles). Persistence after guinea pig absorption but removal by beef absorption = EBV heterophile antibody.
Sensitivity increases with age:
- <4 years: <20%
4 years children: ~40%
- Adults: ~80%
So a negative Monospot in a young child does NOT rule out EBV - use EBV-specific serology.
False positives: acute HIV, rubella, SLE, lymphoma, hepatitis, malaria (~2-3%).
EBV-specific serology (antibodies the patient makes against EBV):
| Acute mono |
+ |
+/- |
+ |
- |
| Recent/convalescent |
+/- |
+ |
+ |
+/- |
| Past infection |
- |
+ |
- |
+ |
| Burkitt reactivation |
- |
+ (high) |
+ (EA-R) |
+ |
| NPC |
- |
+ (high, IgA) |
+ (EA-D) |
+ |
Key points: EBNA appears last (6-12 weeks) and persists for life, so VCA IgM+ / EBNA- = acute infection (most reliable combination). VCA IgG persists lifelong. EA is transient in acute infection; its return suggests reactivation.
Early antigen has two patterns: EA-diffuse (nucleus + cytoplasm, associated with acute mono and NPC, not destroyed by methanol) and EA-restricted (cytoplasm only, associated with Burkitt and chronic/reactivated EBV, destroyed by methanol fixation). NPC is characterized by high VCA IgA reflecting mucosal involvement.
Tissue detection: EBV produces no cytopathic effect in culture - instead it immortalizes B cells into lymphoblastoid cell lines (which is how it was discovered). So culture is not used for diagnosis.
EBER-ISH (EBV-encoded RNA in situ hybridization) is the gold standard for tissue detection. EBER is expressed at 106-107 copies per cell in all latency patterns, making it extremely sensitive. Latency typing by immunohistochemistry:
- Latency I: EBER+, EBNA1 only - Burkitt lymphoma
- Latency II: EBER+, EBNA1 + LMP1, LMP2 - Hodgkin, NPC, NK/T
- Latency III: EBER+, all EBNAs + all LMPs - PTLD, immunodeficiency-associated lymphomas
Quantitative blood EBV PCR is used for monitoring (PTLD risk in transplant, viral load for primary CNS lymphoma in AIDS).
Cytomegalovirus (CMV) is usually asymptomatic in immunocompetent people or causes a mononucleosis-like syndrome. It establishes latency in monocytes and lymphocytes. In immunocompromised patients - AIDS, transplant recipients - CMV reactivation causes retinitis (can cause blindness), colitis, esophagitis, and pneumonitis. Congenital CMV (the most common congenital infection) can cause hearing loss, microcephaly, and developmental delay.
CMV mononucleosis is heterophile-negative (distinguishing it from EBV mono), with less pharyngitis/lymphadenopathy and more hepatitis. Atypical lymphocytes still occur. Seroprevalence ranges from 40-100% depending on population.
CMV disease by host:
- Transplant: CMV is the most important viral pathogen. D+/R- (donor positive, recipient negative) is the highest-risk mismatch for primary disease. Reactivation also common.
- AIDS: disease emerges when CD4 <50 - classic presentations are CMV retinitis (blindness), colitis, esophagitis.
- Congenital CMV: periventricular calcifications, microcephaly, hepatosplenomegaly, jaundice, petechiae (blueberry muffin baby), chorioretinitis, and sensorineural hearing loss (the leading infectious cause of childhood SNHL). Most severe with primary maternal infection in the first trimester.
Histology - the owl eye: CMV produces large basophilic intranuclear inclusions surrounded by a clear halo (owl eye) PLUS smaller granular basophilic cytoplasmic inclusions, all within a dramatically enlarged cell (cytomegaly). The combination of nuclear owl eye + cytoplasmic inclusions + cytomegaly is pathognomonic. CMV does NOT show the Three M’s.
CMV diagnostics:
- Quantitative blood CMV DNA PCR is the standard for diagnosis and monitoring. Rising viral load in transplant patients triggers preemptive therapy.
- Tissue biopsy with owl-eye inclusions confirms end-organ disease.
- Urine or saliva PCR within the first 3 weeks of life diagnoses congenital CMV (after 3 weeks, positive results could represent postnatal acquisition).
- pp65 antigenemia (historical) - stain peripheral leukocytes for the CMV tegument protein. Largely replaced by PCR. Labor-intensive, affected by neutropenia, requires processing within 6 hours.
- Serology: limited utility because seroprevalence is high. Useful for pre-transplant screening (D/R status), diagnosing primary infection by seroconversion or 4-fold IgG rise, or IgG avidity (low avidity = recent primary infection - especially important in pregnancy). CMV IgM alone is unreliable (can persist months; cross-reactivity with EBV, rheumatoid factor).
- Shell vial culture with fluorescent antibody is faster than conventional culture but has lower sensitivity than PCR.
CMV treatment:
- Ganciclovir (IV) / valganciclovir (oral prodrug) - first line. Must be phosphorylated by viral UL97 kinase, then inhibits the CMV DNA polymerase (UL54). Main toxicity: myelosuppression (neutropenia, thrombocytopenia).
- Foscarnet - pyrophosphate analog, directly inhibits UL54 polymerase, does NOT require UL97 activation. Nephrotoxicity, hypocalcemia, hypomagnesemia.
- Cidofovir - nucleotide analog, also UL97-independent. Nephrotoxic.
- Letermovir - CMV terminase inhibitor, used for prophylaxis in HSCT.
- Maribavir - UL97 kinase inhibitor, an option for resistant CMV.
Resistance genetics:
- ==UL97 mutations == cause resistance to ganciclovir only (can’t activate it). Most common resistance mechanism. Switch to foscarnet.
- UL54 mutations (DNA polymerase) can cause resistance to ganciclovir, foscarnet, and/or cidofovir depending on the specific mutation.
- Resistance testing by NGS of UL97/UL54 from blood (requires viral load ≥1000 copies/mL). Indications: rising viral load or clinical progression despite therapy.
HHV-6 and HHV-7 cause roseola (exanthem subitum, sixth disease) in children 6 months to 2 years. Classic pattern: high fever (often >40C) for 3-5 days, then rash as fever resolves. Rose-pink maculopapular rash starts on trunk, spreads peripherally. HHV-6 is the most common cause of febrile seizures in children (~30% of first episodes). In HSCT patients, HHV-6 reactivation (30-50% of recipients, 2-4 weeks post-transplant) can cause limbic encephalitis (altered mental status, seizures, memory impairment) and delay engraftment.
HHV-8 (KSHV) primary infection is usually asymptomatic. Transmission: saliva (major route in endemic areas like sub-Saharan Africa), sexual contact (especially MSM), transplantation. Disease emerges with immunosuppression.
HHV-8 causes three malignancies:
- Kaposi sarcoma - vascular neoplasm with four epidemiologic forms (classic/Mediterranean, endemic/African, iatrogenic/transplant, AIDS-associated). Histology: spindle cells forming slit-like vascular spaces with extravasated RBCs and hemosiderin. Diagnosis: HHV-8 LANA-1 immunohistochemistry (stippled nuclear staining) - distinguishes from bacillary angiomatosis, angiosarcoma, pyogenic granuloma.
- Primary effusion lymphoma (PEL) - B-cell lymphoma presenting as body cavity effusions without a solid mass.
- Multicentric Castleman disease (MCD) - B-cell lymphoproliferative disorder driven by viral IL-6 (vIL-6), presenting with fever, night sweats, lymphadenopathy, cytopenias, elevated CRP. First-line treatment: rituximab.
HHV-8 oncogenesis: LANA (latency-associated nuclear antigen) inactivates p53 and Rb, and HHV-8 encodes viral homologs of cellular genes: vCyclin, vFLIP (anti-apoptotic), vGPCR (constitutively active receptor driving angiogenesis), and vIL-6.
HSV and VZV diagnostic tests: PCR is the most sensitive method for detecting HSV in CSF (for encephalitis) and for typing genital lesions. Direct fluorescent antibody (DFA) and Tzanck smear are less sensitive but faster. The Tzanck smear - scraping the base of a vesicle, staining with Wright or Giemsa, and looking for multinucleated giant cells - can confirm herpesvirus infection but cannot distinguish HSV from VZV. Viral culture demonstrates characteristic cytopathic effect. Type-specific serology (IgG to HSV-1 or HSV-2) is useful for determining prior exposure but not for diagnosing acute lesions.
EBV and infectious mononucleosis diagnosis: The classic triad is fever, pharyngitis, and lymphadenopathy - but what makes the diagnosis is the peripheral blood smear. Atypical lymphocytes are reactive CD8+ T cells responding to EBV-infected B cells. They’re large with abundant basophilic cytoplasm that seems to hug adjacent red cells. The heterophile antibody test (Monospot) detects antibodies that agglutinate sheep or horse red cells - a curious phenomenon caused by polyclonal B cell activation. It’s positive in about 85% of cases but may be negative early in illness or in young children. EBV-specific serology provides definitive diagnosis: VCA-IgM indicates acute infection, VCA-IgG indicates past exposure, and EBNA (Epstein-Barr nuclear antigen) antibodies develop late and indicate past infection.
CMV diagnosis depends on the clinical context: In transplant patients, quantitative PCR (viral load) is the standard for monitoring - rising viral load triggers preemptive therapy. The pp65 antigenemia assay detects CMV protein in peripheral blood leukocytes and was widely used before PCR became standard. Histopathology showing the characteristic “owl’s eye” inclusions - large cells with both nuclear and cytoplasmic inclusions - is diagnostic in tissue specimens.
Hepatitis B Virus (HBV)
Hepadnavirus - an enveloped partially double-stranded DNA virus that replicates via an RNA intermediate using reverse transcriptase. The only DNA hepatitis virus (all others are RNA). After entry, the partially dsDNA genome is converted to covalently closed circular DNA (cccDNA) in the nucleus, which templates viral mRNA and pregenomic RNA. The pregenomic RNA is reverse-transcribed back to DNA by the viral polymerase. This is Baltimore Group VII. The persistent nuclear cccDNA is the reason HBV can never be fully cleared - it persists silently even after apparent recovery, creating reactivation risk with immunosuppression (rituximab, HSCT).
HBV produces three particle types in blood:
- Dane particle (42 nm) - complete infectious virion with genome, polymerase, HBcAg core, enveloped in HBsAg
- Spheres (22 nm) - HBsAg-only particles, non-infectious, outnumber Dane particles 1000:1 (act as immune decoys)
- Filaments/tubules - elongated HBsAg particles, also non-infectious
Key proteins and their locations:
- HBsAg - envelope (surface antigen)
- HBcAg - core nucleocapsid, never free in serum (only detectable by IHC on tissue, nuclear and cytoplasmic staining)
- HBeAg - secreted form of core antigen, free in serum, marker of active replication and high infectivity. Precore G1896A mutants cannot make HBeAg despite active replication.
- HBV polymerase - core, has reverse transcriptase, DNA polymerase, and RNase H activities
- HBx - regulatory transactivator, linked to HCC
Transmission (HBV is 50-100x more infectious than HIV and survives 7+ days on surfaces): sex, blood (transfusion, IVDU, needlestick), vertical (perinatal, most common in endemic areas).
Hepatitis D (delta) is a defective virus that requires HBV for its envelope (HBsAg). Enveloped, negative-sense ssRNA, smallest genome of any animal virus (~1.7 kb). Coinfection (simultaneous HBV + HDV) is usually self-limited. Superinfection of a chronic HBV carrier has high chronicity (~70-80%) and accelerated progression to cirrhosis - the most severe form of viral hepatitis.
Chronicity is inversely related to age at infection:
- Neonates: ~90%
- Children (1-5 years): ~30-50%
- Adults: <10%
Chronic HBV = HBsAg positive for >6 months. Extrahepatic manifestations of chronic HBV include polyarteritis nodosa (immune complex vasculitis), membranous nephropathy (most common glomerular lesion), cryoglobulinemia (type II/III), and aplastic anemia.
Serologic timeline of acute infection:
- HBV DNA - appears first (days after exposure)
- HBsAg - ~4 weeks
- HBeAg - shortly after HBsAg (high replication)
- Anti-HBc IgM - 6-10 weeks, with symptoms
- HBeAg loss and anti-HBe appearance
- HBsAg disappears (~3-4 months)
- Window period (anti-HBc IgM only positive marker)
- Anti-HBs - last, immunity
- Anti-HBc IgG persists lifelong
Serology interpretation (master this table):
| HBsAg |
+ |
+ |
+ |
- |
- |
- |
| Anti-HBs |
- |
- |
- |
- |
+ |
+ |
| Anti-HBc IgM |
+ |
- |
- |
+ |
- |
- |
| Anti-HBc total |
+ |
+ |
+ |
+ |
+ |
- |
| HBeAg |
+ |
+ |
- |
- |
- |
- |
| Anti-HBe |
- |
- |
+ |
+ |
+ |
- |
| HBV DNA |
+ |
high |
low/none |
+/- |
- |
- |
Key diagnostic principles:
- Vaccinated = anti-HBs positive, anti-HBc NEGATIVE (vaccine contains only recombinant HBsAg; no core exposure)
- Natural immunity = anti-HBs positive AND anti-HBc positive
- Anti-HBc IgM is the key marker of acute infection - stays positive through the window period when HBsAg has cleared but anti-HBs hasn’t yet appeared
- HBeAg+ = high infectivity, high viral load (except in precore mutants)
- Isolated anti-HBc positivity (anti-HBc+, HBsAg-, anti-HBs-) has four interpretations: window period, remote past infection with waning anti-HBs, occult low-level chronic HBV, or false positive. Workup: anti-HBc IgM and HBV DNA.
Quantitative HBV DNA is used for treatment monitoring. Current first-line antivirals are entecavir and tenofovir - nucleos(t)ide analogs that inhibit the HBV reverse transcriptase/polymerase. Because HBV RT and HIV RT are targeted by similar drugs, some HIV NRTIs (tenofovir, lamivudine, entecavir) also treat HBV - stopping these in HIV/HBV coinfected patients can cause a severe HBV flare.
Other DNA Viruses
Adenovirus - The Cause of Many Common Syndromes
Adenoviruses are non-enveloped, double-stranded linear DNA viruses with an icosahedral capsid. Characteristic fiber proteins project from the 12 vertices - these bind the Coxsackievirus-Adenovirus Receptor (CAR) on host cells for attachment. Over 70 serotypes exist, and different serotypes have tropism for different tissues. Being non-enveloped makes adenovirus environmentally resistant, enabling both respiratory and fecal-oral transmission.
Serotype to syndrome (high yield):
- Serotypes 1-7: respiratory infections (pharyngitis, pneumonia). Types 3, 4, 7, 14 most common.
- Serotypes 8, 19, 37: epidemic keratoconjunctivitis (EKC)
- Serotypes 11 and 21: hemorrhagic cystitis
- Serotypes 40 and 41: enteric, cause gastroenteritis
- Serotypes 1, 2, 5: disseminated disease in immunocompromised (especially HSCT)
Respiratory disease: Adenovirus causes pharyngitis, often with conjunctivitis (pharyngoconjunctival fever), and can cause severe pneumonia, particularly in military recruits, infants, and immunocompromised patients. A live oral vaccine for types 4 and 7 is used in the military and has dramatically reduced outbreaks in crowded barracks.
Epidemic keratoconjunctivitis: highly contagious, bilateral (starts unilateral, spreads within days), watery discharge with preauricular lymphadenopathy. Subepithelial corneal infiltrates can develop 1-3 weeks later (immune-mediated, can persist months). Classically spreads through ophthalmology clinics via tonometers and hands, and through swimming pools.
Hemorrhagic cystitis: gross hematuria, dysuria. Other causes to remember: BK virus, cyclophosphamide/ifosfamide. Adenovirus hemorrhagic cystitis is usually self-limiting in immunocompetent hosts but can be severe and prolonged in transplant patients.
Gastroenteritis: enteric serotypes 40 and 41 are the second most common viral cause of childhood diarrhea after rotavirus. Adenovirus and rotavirus diarrhea can both precipitate intussusception via mesenteric lymphoid hyperplasia.
Disseminated disease in immunocompromised (especially HSCT, pediatric solid organ transplant): hepatitis (most common organ), pneumonitis, colitis, hemorrhagic cystitis, encephalitis. Mortality 50-80% in HSCT. Monitor with weekly blood adenovirus PCR. Treatment: cidofovir or brincidofovir, and reduce immunosuppression.
Diagnostics:
- PCR is the primary method - often part of multiplex respiratory or GI panels (e.g., BioFire FilmArray). Quantitative blood PCR for monitoring viremia in transplant.
- Culture: most serotypes grow on A549, HEK-293, or HeLa over 5-10 days. CPE looks like grape-like clusters of rounded, refractile cells. Serotypes 40 and 41 do not grow in standard culture - they need Graham 293 cells, which is why enteric adenovirus was underdiagnosed before PCR.
- Histology: smudge cells - intranuclear inclusions with basophilic, homogeneous smudged chromatin that obliterates nuclear detail. Early lesions can show “two-toned” nuclei (eosinophilic center with basophilic rim). Seen in lung, liver, and bladder. No cytoplasmic inclusions, no syncytia.
- DFA and rapid antigen: ~50-70% sensitivity, inferior to PCR.
Parvovirus B19 - The Red Cell Specialist
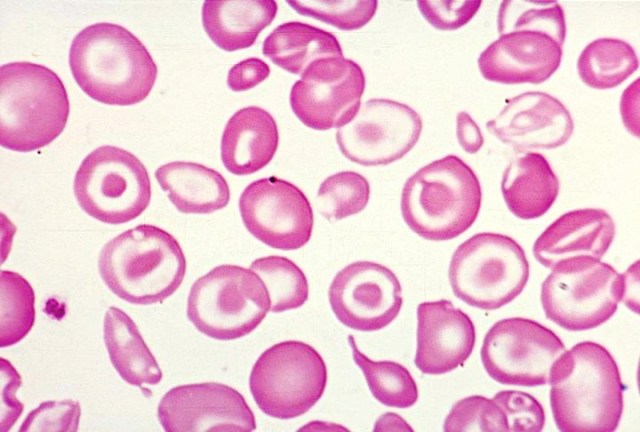
Parvovirus B19 is a small, non-enveloped, single-stranded DNA virus (the smallest DNA virus infecting humans) with a remarkably specific tropism: it infects and destroys erythroid progenitor cells. This tropism explains all of its clinical manifestations. Because it’s non-enveloped, it is NOT inactivated by solvent/detergent treatment of blood products - hence transfusion-transmitted risk. It requires dry heat or nanofiltration (15 nm) for inactivation.
Pathogenesis: Parvovirus B19 binds to the P antigen (globoside, Gb4) on red blood cell precursors in the bone marrow. Individuals with the rare p phenotype (lacking P antigen) are naturally resistant to parvovirus B19. The same P antigen is the target of the Donath-Landsteiner antibody in paroxysmal cold hemoglobinuria. The virus replicates in the nucleus of normoblasts/proerythroblasts and kills them as new virions are released. In normal individuals with healthy bone marrow, this destruction causes a temporary reticulocytopenia lasting 7-10 days, but the long lifespan of circulating RBCs (120 days) means this goes unnoticed - the marrow recovers before anemia develops.
Epidemiology: peak incidence at ages 3-7 years. Outbreaks occur in late winter and spring, with epidemics every 3-7 years. Patients are most infectious before the rash appears - by the time the slapped cheek rash is visible, viremia has cleared and the child is no longer contagious.
Erythema infectiosum (fifth disease) is the classic childhood illness - a mild febrile illness followed by the “slapped cheek” rash (bright red erythema of the cheeks) and then a lacy, reticular rash on the trunk and extremities. The rash represents the immune response to the virus and appears as viremia clears; by the time the rash appears, the child is no longer contagious.
Aplastic crisis occurs when parvovirus B19 infects someone with underlying hemolytic anemia (sickle cell disease, hereditary spherocytosis, thalassemia). These patients depend on accelerated red cell production to compensate for their shortened RBC lifespan. When parvovirus halts erythropoiesis, their hemoglobin plummets precipitously - a potentially life-threatening aplastic crisis requiring transfusion.
Hydrops fetalis occurs when a seronegative pregnant woman is infected during pregnancy. The fetus lacks immunity and cannot clear the virus. Massive destruction of fetal red cell precursors causes severe fetal anemia, leading to high-output heart failure, ascites, and hydrops. In severe cases, fetal death results. Intrauterine transfusion can be life-saving.
Chronic anemia in immunocompromised patients occurs because they cannot mount the antibody response needed to clear the virus. Persistent infection causes ongoing pure red cell aplasia and severe, chronic anemia requiring transfusions. Treatment with IVIG provides anti-parvovirus antibodies that clear the infection.
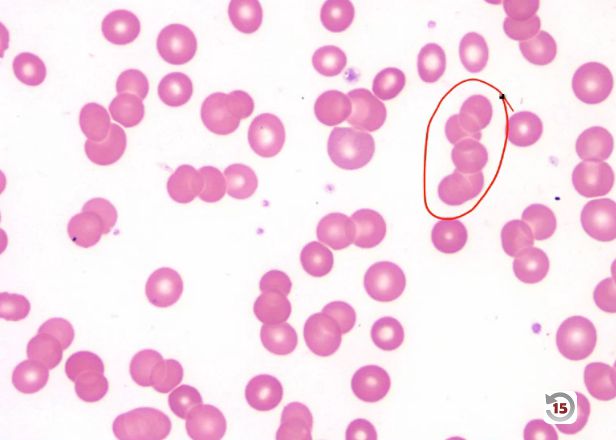
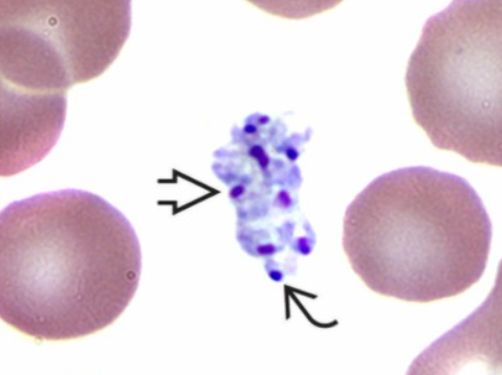
Bone marrow morphology is pathognomonic: giant pronormoblasts (“lantern cells”) with glassy eosinophilic nuclear inclusions surrounded by a clear zone, and absent downstream erythroid maturation. Diagnosis can also be made by PCR of blood or bone marrow (most sensitive), immunohistochemistry for VP1/VP2 capsid proteins, or in situ hybridization.
Serology: IgM appears first (persists 2-3 months, indicates acute); IgG confers lifelong immunity. Interpretation: IgM+/IgG- = acute; IgM+/IgG+ = recent; IgM-/IgG+ = past/immune. Hydrops fetalis occurs only with primary maternal infection.
Human Papillomavirus (HPV) - Warts and Cancer
HPV is a diverse family of over 200 genotypes that infect cutaneous and mucosal squamous epithelium. The clinical significance varies dramatically by type: some cause benign warts, while others cause cancer. Understanding the molecular basis of HPV-driven carcinogenesis explains why vaccines targeting high-risk types are so important. HPV is a non-enveloped, circular double-stranded DNA virus. HPV cannot be grown in standard cell culture (requires differentiating epithelium), so diagnosis is molecular/histologic.
Two infection patterns - master this distinction:
- Permissive (episomal) infection - viral DNA stays circular and non-integrated, active virion production, cell lysis. E2 protein intact, suppressing E6/E7. Result: benign warts and condylomata. Associated with low-risk types (6, 11).
- Non-permissive (integrated) infection - viral DNA integrates into host chromosome. Integration disrupts E2, releasing E6/E7 from suppression. Overexpression of E6/E7 drives dysplasia and cancer. Associated with high-risk types (16, 18, 31, 33, 45).
Low-risk types (6 and 11) cause genital warts (condyloma acuminata) - exophytic, cauliflower-like lesions on the anogenital epithelium. These types do not cause cancer but cause significant morbidity. They can also cause laryngeal papillomatosis when transmitted to neonates during delivery.
High-risk types (especially 16 and 18) are oncogenic. These types are responsible for the vast majority of cervical cancers, a substantial proportion of oropharyngeal (tonsil, base of tongue) cancers, and anal cancers. The mechanism of carcinogenesis involves two viral oncoproteins:
E6 protein binds to and promotes degradation of p53, the tumor suppressor that normally arrests the cell cycle when DNA damage is detected. Without p53, damaged cells continue to proliferate.
E7 protein inactivates the retinoblastoma protein (Rb), which normally prevents cells from entering S phase inappropriately. With Rb inactivated, cells divide uncontrollably.
Together, E6 and E7 override the two major tumor suppressor pathways, allowing accumulation of mutations and progression to cancer over years to decades of persistent infection.
The vaccine targets the capsid proteins of high-risk types (16, 18, and others in the 9-valent vaccine) and low-risk types (6, 11). By preventing infection with these types, the vaccine prevents the cancers and warts they cause - one of the few vaccines that prevents cancer.
HPV type to disease (memorize the mappings):
- Common/plantar warts (verruca vulgaris) - HPV 1, 2, 4 (plus 7 for butcher warts)
- Verruca plana (flat warts) - HPV 3, 10
- Genital warts (condyloma acuminata) - HPV 6, 11 (low-risk)
- Recurrent respiratory papillomatosis (laryngeal papillomatosis) - HPV 6, 11. Juvenile-onset acquired from infected birth canal; adult-onset sexually acquired.
- Cervical HSIL/SCC and adenocarcinoma - HPV 16, 18 (plus 31, 33, 45, 52, 58). HPV 16 is most common in cervical SCC; HPV 18 is most common in cervical adenocarcinoma. Together 16+18 cause ~70% of cervical cancers.
- HPV 16 also the dominant driver of oropharyngeal (tonsil, base of tongue) SCC, anal SCC, vulvar/vaginal/penile SCC.
Epidermodysplasia verruciformis (EV): autosomal recessive disease with TMC6/TMC8 (EVER1/EVER2) mutations conferring susceptibility to HPV 5 and 8. Widespread flat wart-like lesions and pityriasis versicolor-like plaques in sun-exposed areas; 30-60% develop SCC by age 40-50. Histology shows characteristic bluish-gray cytoplasmic staining of keratinocytes (viral protein accumulation) and prominent keratohyalin granules.
Diagnostics and IHC:
- High-risk HPV shows block-positive p16 staining (diffuse strong nuclear + cytoplasmic). Mechanism: E7-mediated Rb inactivation drives compensatory p16 overexpression. Used for CIN2+, HPV-associated oropharyngeal SCC.
- Cervical HPV testing: hrHPV molecular testing of cervical cytology specimens detects the 14 high-risk types. Cotest (cytology + hrHPV) or primary HPV testing are accepted screening paradigms.
- Koilocytes - perinuclear halos with nuclear atypia - are the classic cytologic finding in productive infection.
Human Polyomaviruses - JC, BK, and Merkel
The human polyomaviruses are small, non-enveloped, circular dsDNA viruses. Primary infection occurs in childhood (usually asymptomatic, seroprevalence 70-90% by adulthood), with lifelong latency. Disease emerges with immunosuppression. The name “polyoma” comes from their ability to cause multiple tumors in experimental animals. Their early gene region encodes large T and small t antigens that interact with p53 and Rb - directly relevant to their oncogenic potential.
Three clinically important human polyomaviruses: JC virus (PML), BK virus (nephropathy, hemorrhagic cystitis), and Merkel cell polyomavirus (Merkel cell carcinoma).
JC Virus - The Demyelinating Polyomavirus
JC virus (named for John Cunningham, the patient from whom it was first isolated) is a polyomavirus that infects most people asymptomatically in childhood and establishes latent infection in the kidneys. In profoundly immunocompromised patients - particularly those with AIDS or on certain immunosuppressive therapies like natalizumab (for multiple sclerosis), rituximab, or other monoclonal antibodies - the virus can reactivate and cause progressive multifocal leukoencephalopathy (PML).
PML pathogenesis: JC virus reactivates and travels to the central nervous system, where it infects oligodendrocytes - the cells that produce myelin. Multifocal areas of demyelination appear throughout the brain. JCV also infects astrocytes, producing bizarre, pleomorphic reactive astrocytes on histology. The clinical presentation is subacute neurological decline with cognitive impairment, visual disturbances, weakness, and ataxia.
MRI and CSF findings - memorable because they’re “quiet”:
- MRI: non-enhancing, non-mass-forming white matter lesions with T2/FLAIR hyperintensity, predilection for parieto-occipital regions. U-fibers involved (unlike MS). No ring enhancement (unlike toxoplasmosis or CNS lymphoma).
- CSF: typically normal or near-normal glucose, protein, and cell count. This “bland” CSF contrasts with bacterial meningitis and viral encephalitis.
- CSF JCV PCR - sensitivity 72-92%, specificity >99%. Negative PCR does not exclude PML.
- Histology: enlarged oligodendrocytes with ground-glass nuclear inclusions, foamy macrophages.
PML is almost always fatal without immune reconstitution. In AIDS patients, antiretroviral therapy can restore immunity. In patients on immunosuppressive therapy, discontinuing the offending agent is essential.
BK Virus - The Kidney Transplant Threat
BK virus establishes latent infection in the kidneys (renal tubular epithelium, urothelium) and CNS after childhood acquisition. Low-level BK viruria is present in 5-10% of healthy immunocompetent adults. Disease emerges primarily in transplant recipients.
BK nephropathy (BKVN) occurs in 1-10% of kidney transplants. Tacrolimus > cyclosporine for risk; ureteral stent placement is another risk factor. The virus infects tubular epithelial cells, causing tubular necrosis, interstitial inflammation (which can mimic acute rejection), and potentially graft loss. Distinguishing BKVN from rejection is critical because treatment is opposite: reduce immunosuppression for BK, increase for rejection.
BK hemorrhagic cystitis occurs mainly in HSCT recipients (allogeneic > autologous).
Diagnostics:
- Urine BK PCR - sensitive screening; viruria precedes viremia by weeks.
- Plasma BK PCR - more specific for clinically significant disease. Plasma BK >10,000 copies/mL is presumptive BKVN - biopsy recommended. Monitoring protocol in renal transplant: urine BK PCR monthly x 6 months, then every 3 months for 2 years.
- Urine cytology: decoy cells - enlarged nuclei with ground-glass intranuclear inclusions that mimic high-grade urothelial carcinoma. Clues to viral origin: smooth nuclear membranes, homogeneous chromatin.
- Kidney biopsy: tubular epithelial cells with enlarged nuclei containing basophilic ground-glass intranuclear inclusions.
- SV40 IHC - the gold-standard tissue stain. The anti-SV40 large T antigen antibody cross-reacts with both BK and JC virus (~70% homology). Cannot distinguish the two - context determines which.
Treatment: reduce immunosuppression. No proven antiviral. IVIG and cidofovir have been tried but are not standard of care.
Merkel Cell Polyomavirus (MCPyV)
MCPyV is clonally integrated in ~80% of Merkel cell carcinomas - aggressive neuroendocrine skin tumors of elderly, immunosuppressed, sun-exposed patients. The MCPyV large T antigen inactivates Rb (similar to HPV E7); small T antigen inhibits PP2A. Histology: small blue round cells with neuroendocrine features. IHC: CK20 paranuclear dot pattern, synaptophysin, chromogranin positive. MCPyV has also been associated with CLL/SLL.
TORCH Infections: Congenital Infection Overview
TORCH (or TORCHES) is the classic mnemonic for pathogens causing congenital infection. These infections can have devastating consequences for the fetus and newborn.
| Toxoplasma |
Intracranial calcifications (diffuse), chorioretinitis, hydrocephalus |
Serology, PCR |
| Other (Syphilis, VZV, Parvovirus B19) |
Syphilis: snuffles, rash, bone abnormalities; Parvovirus: hydrops fetalis |
Serology |
| Rubella |
Cataracts, congenital heart defects (PDA), sensorineural deafness, “blueberry muffin” rash |
Serology (IgM) |
| CMV |
Periventricular calcifications, sensorineural hearing loss, microcephaly |
Urine PCR (shell vial culture) |
| HSV |
Skin vesicles, encephalitis, disseminated disease; often acquired during delivery |
PCR, culture |
| EBV |
Rare congenital infection |
- |
| Syphilis |
See “Other” above |
RPR/VDRL, FTA-ABS |
Key distinguishing feature: Toxoplasma causes diffuse/scattered intracranial calcifications; CMV causes periventricular calcifications. This is a classic boards distinction.
CMV is the most common congenital infection and the leading infectious cause of sensorineural hearing loss in children.
49.2 RNA Viruses
Picornaviruses: Small RNA Viruses with Big Impact
The Picornaviridae - the name means “small RNA viruses” - are among the simplest viruses that infect humans, yet they cause an enormous range of disease. They’re non-enveloped (naked) positive-sense ssRNA viruses, which makes them resistant to environmental conditions, detergents, and drying. They’re acid-stable, allowing them to survive passage through the stomach. Being positive-sense means their genome can be immediately translated by host ribosomes upon entry - no need for conversion or transcription first. They’re icosahedral, about 30 nm.
Clinically important picornaviruses:
- Enteroviruses: poliovirus, coxsackievirus A and B, echovirus, enterovirus D68, enterovirus 71. Parechovirus is grouped here for board purposes.
- Rhinovirus: most common cause of common cold
- Hepatitis A (Hepatovirus): fecal-oral acute hepatitis
Enteroviruses cause ~75% of viral (aseptic) meningitis with peak incidence in summer and early fall. CSF profile: lymphocytic pleocytosis, normal glucose, normal to mildly elevated protein. Diagnosis: CSF enterovirus PCR.
Poliovirus - The Vaccine Success Story
Poliomyelitis has been virtually eliminated from the developed world through vaccination - one of medicine’s great triumphs. But understanding the disease remains important, both historically and because polio persists in some regions and remains a bioterrorism concern.
Pathogenesis explains the clinical spectrum: Poliovirus enters through the mouth, replicates in the oropharynx and gut-associated lymphoid tissue (particularly Peyer’s patches), and is shed in feces. In most people (95%), infection stops here - asymptomatic or a nonspecific febrile illness. In some, viremia carries the virus to the central nervous system.
The critical target is the anterior horn motor neurons of the spinal cord. The virus preferentially infects and destroys these cells. Because motor neurons are post-mitotic - they can’t regenerate - destruction is permanent. The result is lower motor neuron disease: asymmetric flaccid paralysis with muscle atrophy, fasciculations, and absent reflexes. Sensation is preserved because sensory neurons are spared. This constellation - asymmetric flaccid paralysis with intact sensation - is classic for polio and distinguishes it from Guillain-Barré syndrome (which affects peripheral nerves, causing sensory and motor findings).
Bulbar polio, involving cranial nerve motor nuclei, causes dysphagia and respiratory failure. The iron lung - an image that defines mid-20th century polio epidemics - provided artificial ventilation for patients with respiratory muscle paralysis.
Two vaccines, two strategies: Jonas Salk developed the inactivated polio vaccine (IPV), which uses killed virus given by injection. It induces systemic IgG antibodies that prevent viremia and thus paralytic disease - but it doesn’t induce mucosal immunity, so vaccinated individuals can still carry and transmit the virus.
Albert Sabin developed the oral polio vaccine (OPV), which uses live attenuated virus. Given orally, it replicates in the gut, inducing both systemic IgG and mucosal IgA. This prevents both disease and transmission. However, because it’s a live virus, OPV can rarely revert to virulence, causing vaccine-associated paralytic polio (VAPP, ~1 per 2.4 million doses). The US now uses only IPV; OPV remains important in global eradication campaigns because it’s cheaper, easier to administer, and induces mucosal immunity.
Memory hook: Salk is sulking because he’s dead (Salk = killed/inactivated, IM); Sabin = oral, live.
Coxsackievirus - The Heart and Hand Virus
Coxsackieviruses are divided into groups A and B based on their effects in newborn mice - but more importantly, based on the different clinical syndromes they cause.
Coxsackie A has tropism for skin and mucous membranes. Classic diseases:
- Hand-foot-mouth disease (HFMD) - usually Coxsackie A16 or Enterovirus 71, vesicles on hands, feet, and oral mucosa.
- Herpangina - painful vesicles/ulcers on the posterior oropharynx (soft palate, uvula, tonsils). Distinguishes from HSV gingivostomatitis, which affects the anterior mouth.
- Acute hemorrhagic conjunctivitis
- Aseptic meningitis
Coxsackie B has tropism for heart, pleura, and pancreas (“B is for Body”). Classic diseases:
- Viral myocarditis and pericarditis - Coxsackie B is the most common viral cause. Can progress to dilated cardiomyopathy.
- Pleurodynia (Bornholm disease) - sudden severe pleuritic chest pain, often the worst pain of the patient’s life. Self-limited but mimics MI/PE.
- Aseptic meningitis
Both Coxsackie A and B can cause aseptic meningitis. Enterovirus culture (Rhesus monkey kidney, MRC-5, Vero, or rhabdomyosarcoma cells) takes 4-8 days with rounded refractile degenerating CPE but has been largely replaced by PCR.
Enterovirus D68 caused a 2014 US outbreak of severe respiratory illness in children (especially asthmatics) and was linked to acute flaccid myelitis, a polio-like syndrome with anterior horn cell damage. Enterovirus 71 causes HFMD with CNS complications.
Rhinovirus and Echovirus: Rhinovirus is the most common cause of the common cold (~50-80% of colds), with over 160 serotypes (making vaccination impractical). Replicates best at 33°C, the cooler temperature of the nasal mucosa, which is why it sticks to the upper airways. Transmitted by aerosols and fomites; survives on surfaces for hours. Can trigger asthma exacerbations. Diagnosis is by multiplex PCR, which often cannot distinguish rhinovirus from enterovirus due to sequence homology - they may be reported together as “rhinovirus/enterovirus.”
Echovirus (Enteric Cytopathic Human Orphan virus - “orphan” because initially no disease was linked) is a major cause of aseptic meningitis, with >30 serotypes. Also causes rash, conjunctivitis, respiratory illness, and severe neonatal sepsis-like illness.
Hepatitis A
A picornavirus (Hepatovirus) transmitted by fecal-oral route - contaminated water, shellfish, produce, person-to-person. Crowded settings in the US: daycares are a classic reservoir (children are often asymptomatic shedders who transmit to adults). Recent outbreaks: people who use drugs, homeless populations, MSM.
Natural history:
- Incubation 2-6 weeks
- Symptomatic acute hepatitis: jaundice, fever, malaise, transaminases often >1000
- Usually lasts <2 months
- NEVER becomes chronic. No carrier state.
- ~5% have a relapsing course
- Fulminant hepatic failure <1%
- Severity increases with age (adults more often symptomatic than children)
Diagnosis and prevention: Anti-HAV IgM indicates acute infection; anti-HAV IgG indicates past infection or vaccination and confers lifelong immunity.
The hepatitis A vaccine is killed (inactivated) whole-virus, two doses 6-12 months apart, >95% effective. Recommended for all children at age 1, travelers to endemic areas, MSM, people who use drugs, patients with chronic liver disease, homeless populations. Post-exposure prophylaxis: HAV vaccine preferred for healthy adults 1-40; immunoglobulin for infants, immunocompromised, older adults.
Paramyxoviruses: Respiratory Viruses with Fusion
The paramyxoviruses are large, enveloped, negative-sense ssRNA, non-segmented viruses that share a key feature: a fusion (F) protein that allows them to spread directly from cell to cell, creating multinucleated giant cells (syncytia). This cell-to-cell spread helps them evade antibodies in the extracellular space.
Because paramyxoviruses have a non-segmented genome, they do not undergo reassortment - no antigenic shift, which is why they cause epidemics but not pandemics (unlike influenza).
Family members:
- Parainfluenza (types 1-4)
- Mumps
- Measles (Morbillivirus)
- RSV (Orthopneumovirus)
- Human metapneumovirus (hMPV)
- Nipah and Hendra (Henipavirus, emerging zoonotic encephalitis from bats)
Measles (Rubeola) - One of the Most Contagious Diseases
Measles is extraordinarily contagious - an infected person can transmit the virus to 90% of susceptible contacts. Before vaccination, virtually everyone was infected in childhood. The virus spreads via respiratory droplets and aerosols, initially infecting respiratory epithelium and then spreading to lymphoid tissue. Viremia disseminates the virus to skin, respiratory tract, and other organs.
The clinical course is predictable and distinctive: After a 10-14 day incubation period, prodromal illness begins with the “3 Cs” plus Koplik spots - Cough, Coryza (runny nose), Conjunctivitis, and Koplik spots. Koplik spots are pathognomonic: tiny white/bluish spots on an erythematous base, appearing on the buccal mucosa opposite the molars. They look like “grains of salt on a red background” and appear 1-2 days before the rash.
The rash emerges as the fever peaks, starting on the face and spreading downward (cephalocaudal) over 3-4 days. It’s maculopapular, eventually becoming confluent. As the rash spreads downward, it begins fading on the face. The rash represents immune-mediated clearing of infected cells - immunocompromised patients may have no rash yet severe, disseminated infection.
Complications explain why measles matters: In developed countries with good nutrition, most cases resolve uneventfully. But complications can be devastating. Pneumonia - primary viral or secondary bacterial - is the most common cause of death. Encephalitis occurs in about 1 in 1000 cases. Most terrifying is subacute sclerosing panencephalitis (SSPE), a progressive, fatal neurodegenerative disease that appears 7-10 years after measles infection, caused by persistence of defective virus in the brain. Presentation: behavioral changes, myoclonic jerks, seizures, progressive cognitive decline. EEG shows periodic high-amplitude complexes; CSF has elevated measles antibody titers and oligoclonal bands. Median survival is 1-3 years after symptom onset.
Histology: Warthin-Finkeldey giant cells are pathognomonic - large multinucleated cells with up to 100 nuclei in lymphoid tissue (tonsils, lymph nodes, appendix, Peyer patches, thymus) representing virus-induced lymphocyte fusion. Can appear before the rash. Unlike HSV/VZV/CMV, measles is the only common virus with BOTH nuclear AND cytoplasmic inclusions.
Diagnosis: PCR of throat/nasopharyngeal swab or urine (most sensitive). Measles IgM appears with the rash; 4-fold rise in IgG between acute and convalescent sera confirms. Measles is a reportable disease - contact public health on suspicion. Airborne + contact isolation.
Vitamin A supplementation reduces measles complications and mortality, particularly in malnourished children. This is one of the most effective and cheapest interventions in global health.
Measles IgM Serology Caveats: IgM may be negative in first 72 hours of rash onset (false-negative window). If clinical suspicion is high, repeat in 3-5 days. Prozone effect (antibody excess) can cause false-negative results. False-positive IgM can occur with EBV infection and rheumatoid factor.
Mumps - The Swollen Gland Virus
Mumps was once a childhood rite of passage; vaccination has made it rare. The virus infects the upper respiratory tract, spreads via viremia, and has particular tropism for glandular and nervous tissue.
Parotitis is the hallmark: Painful swelling of the parotid glands, bilateral in about 70% of cases, gives patients the classic “chipmunk cheeks” appearance. The swelling obscures the angle of the jaw. The parotid duct opening (Stensen’s duct) may be red and edematous.
Orchitis is the feared complication in adult males: In post-pubertal males, mumps virus can infect the testes, causing painful swelling, usually unilateral. The testis becomes edematous within a confined capsule (the tunica albuginea), leading to pressure necrosis. Although orchitis causes testicular atrophy in about 50% of affected testes, sterility is rare because it’s usually unilateral and the contralateral testis compensates.
Aseptic meningitis occurs commonly but is usually benign. Pancreatitis and oophoritis are less common complications.
Parainfluenza - The Croup Virus
Parainfluenza viruses are the most common cause of croup (laryngotracheobronchitis), a distinctive syndrome seen in children 6 months to 3 years old. The subglottic area - just below the vocal cords - is the narrowest part of a child’s airway. When parainfluenza infection causes inflammation and edema here, the airway narrows further, causing the classic symptoms: inspiratory stridor (the high-pitched sound of air passing through a narrowed extrathoracic airway), a “barking” cough (compared to a seal’s bark), and hoarseness.
The X-ray shows the “steeple sign” - the normally squared-off subglottic area appears tapered, like a church steeple, due to edema. Treatment involves corticosteroids (to reduce inflammation) and nebulized epinephrine (to cause vasoconstriction and reduce edema) in moderate-severe cases.
RSV (Respiratory Syncytial Virus) is the leading cause of lower respiratory tract infection in infants, particularly bronchiolitis (infection of the small airways). RSV causes epidemics every winter. In most children, it causes cold-like symptoms, but in young infants - especially premature infants - it can cause severe disease requiring hospitalization. The F (fusion) glycoprotein mediates both entry and cell-cell fusion, creating the syncytia that give the virus its name.
Culture uses HEp-2 cells (human laryngeal carcinoma) - the optimal line for RSV - with characteristic syncytial CPE; sensitivity is ~50-70%, so PCR or rapid antigen has replaced it. Histology shows cytoplasmic inclusions and syncytia with no nuclear inclusions.
RSV prophylaxis (treatment is supportive; inhaled ribavirin in severe immunocompromised cases):
- Palivizumab (Synagis) - monoclonal antibody against F protein, monthly IM during RSV season. For premature infants, chronic lung disease, hemodynamically significant CHD.
- Nirsevimab (Beyfortus) - long-acting monoclonal, single dose covers the whole season.
- Maternal RSV vaccine (Abrysvo) given during pregnancy protects neonates.
Parainfluenza: the leading cause of croup (laryngotracheobronchitis) in children 6 months to 3 years. Types 1 and 2 cause croup; type 3 causes bronchiolitis/pneumonia in infants. Barking cough, inspiratory stridor, steeple sign on X-ray. Treatment: corticosteroids (dexamethasone) and nebulized epinephrine for moderate-severe cases.
Mumps classically targets parotid glands, pancreas, gonads, and CNS:
- Parotitis (bilateral in ~70%) - most common manifestation
- Orchitis in post-pubertal males (~30%, usually unilateral - sterility rare)
- Oophoritis (~5%)
- Pancreatitis (~5%)
- Aseptic meningitis (~10%), encephalitis rarely
Prevention: MMR. Diagnosis: PCR of saliva/buccal swab; IgM serology.
Human metapneumovirus (hMPV) - described in 2001, causes RSV-like illness (bronchiolitis, pneumonia, croup) in children, elderly, and immunocompromised. Peak late winter/spring (slightly later than RSV). Detection: PCR on multiplex respiratory panels. No antiviral, no vaccine.
Rabies Virus (Rhabdovirus) - The Fatal Encephalitis
Rabies is almost unique among infectious diseases: once symptoms appear, it’s essentially 100% fatal. Fewer than 20 people have ever survived symptomatic rabies. This makes prevention - through animal control, vaccination, and post-exposure prophylaxis - absolutely critical.
The virus is a bullet-shaped, enveloped, negative-sense RNA virus (rhabdo = rod/bullet in Greek), ~75 x 180 nm. The envelope is studded with G protein spikes that bind neuronal nicotinic acetylcholine receptors, NCAM, and p75NTR.
Reservoirs:
- United States (>90% wildlife): bats, raccoons, skunks, foxes. Bats are the most common source of human rabies and bat bites may be unrecognized.
- Worldwide: dogs (>95% of human rabies deaths, primarily Asia and Africa, mostly children). ~59,000 annual global deaths.
Pathogenesis explains the long incubation and fatal outcome: After a bite, the virus replicates locally in muscle tissue, then enters peripheral nerves. It travels retrograde along axons toward the central nervous system - a process that can take weeks to months depending on the bite’s distance from the brain. Once the virus reaches the CNS, it replicates explosively in neurons, particularly in the limbic system, hippocampus, and brainstem. By the time symptoms appear, the brain is extensively infected.
The clinical course is harrowing: The prodrome begins with paresthesias or pain at the bite site (often the first clue) - this reflects viral replication in dorsal root ganglia. Fever, malaise, and anxiety develop.
Furious rabies (80% of cases) is the classic form. Patients become agitated, confused, and hyperactive. The pathognomonic symptom is hydrophobia - painful spasms of the pharynx and diaphragm triggered by attempts to swallow, or even by the sight or sound of water. Aerophobia (spasms triggered by drafts of air) is similar. Hypersalivation is prominent; the combination of hypersalivation and inability to swallow produces the “foaming at the mouth” image. Periods of lucidity alternate with periods of agitation.
Paralytic rabies (20% of cases) presents as ascending paralysis mimicking Guillain-Barré syndrome - a diagnostic pitfall.
Both forms progress to coma and death, usually from respiratory failure, within 2 weeks of symptom onset.
Negri bodies - round-to-oval, eosinophilic cytoplasmic inclusions in neurons, particularly in hippocampal pyramidal cells and cerebellar Purkinje cells - are the histologic hallmark but present in only ~70-80% of cases, so absence does not exclude rabies. At autopsy, the brain may appear grossly normal; microscopically, perivascular lymphocytic cuffing and microglial nodules (Babes nodules) accompany the Negri bodies. The gold-standard diagnostic test is direct fluorescent antibody (DFA) on brain tissue at autopsy, or nuchal skin biopsy (containing hair follicles with nerve endings) for antemortem diagnosis.
Post-exposure prophylaxis is life-saving: The long incubation period provides a window for intervention. Wound cleaning with soap and water is critical - physical removal of virus reduces inoculum. Rabies immunoglobulin (RIG) provides immediate passive immunity and should be infiltrated around the wound. The rabies vaccine induces active immunity over subsequent weeks. Given promptly and correctly, PEP is virtually 100% effective.
Pre-exposure vaccination is recommended for high-risk groups: veterinarians, animal handlers, laboratory workers, cave explorers (bat exposure), and travelers to endemic areas where dog rabies is common and access to PEP might be limited.
Influenza
Influenza is an orthomyxovirus with a segmented RNA genome - a feature that enables the genetic reassortment responsible for pandemic emergence. The virus causes annual epidemics and occasional pandemics with massive global impact. Understanding how influenza’s virulence factors work at the molecular level explains its pathogenesis, transmission, and treatment.
The Two Key Surface Glycoproteins - How They Function
Two surface glycoproteins - hemagglutinin (HA) and neuraminidase (NA) - are not just immunologic targets; they are the functional machinery that allows the virus to infect cells, replicate, and spread. Understanding their mechanisms is essential.
Hemagglutinin (HA) - The Key to Entry: Hemagglutinin is a trimeric spike protein that mediates two critical steps in infection. First, HA binds to sialic acid (neuraminic acid) residues on the surface of respiratory epithelial cells. This receptor-binding function initiates infection - without it, the virus cannot attach to target cells. The specific sialic acid linkage matters: human influenza viruses preferentially bind α-2,6-linked sialic acid (found predominantly in human upper respiratory tract), while avian influenza viruses prefer α-2,3 linkages (found in avian intestinal and respiratory epithelia and in human lower respiratory tract). This receptor specificity determines species tropism and transmission efficiency.
Second, after the virus is internalized by endocytosis, HA mediates membrane fusion. As the endosome acidifies, HA undergoes a dramatic conformational change that exposes a fusion peptide, which inserts into the endosomal membrane and pulls the viral and cellular membranes together. This releases the viral genome into the cytoplasm. The HA must be cleaved by host proteases into HA1 and HA2 subunits for this fusion mechanism to work - this cleavage requirement is another determinant of virulence. Highly pathogenic avian influenza strains (like H5N1) have polybasic cleavage sites that can be cleaved by ubiquitous proteases, allowing systemic infection; low-pathogenic strains require trypsin-like proteases found only in the respiratory tract, limiting infection to the airways.
Neuraminidase (NA) - The Key to Release and Spread: Neuraminidase is a tetrameric enzyme that cleaves sialic acid residues. Its function becomes critical at the end of the viral life cycle. Newly synthesized viral particles bud from the infected cell’s surface, but HA on these new virions immediately binds back to sialic acid on the same cell surface - the virus essentially sticks to itself. Neuraminidase solves this problem by cleaving the sialic acid residues, releasing new virions to spread and infect more cells.
Neuraminidase also helps the virus penetrate respiratory mucus (which is rich in sialic acid-containing glycoproteins) to reach epithelial cells. Without functional NA, newly formed virions clump together on the cell surface and cannot disseminate. This is exactly why neuraminidase inhibitors (oseltamivir, zanamivir) are effective antivirals - by blocking NA activity, they prevent viral release and spread, limiting the infection if given early enough (within 48 hours, before the infection is too established).
How Influenza Causes Disease: The pathogenesis of influenza involves direct viral damage and immunopathology. The virus infects and kills ciliated respiratory epithelial cells, disrupting the mucociliary escalator that normally clears pathogens and debris. This denudation of the respiratory epithelium creates susceptibility to secondary bacterial pneumonia - historically the major cause of death in influenza pandemics. The host immune response - pro-inflammatory cytokines including IL-6, TNF-α, and interferons - causes the systemic symptoms of fever, myalgia, and malaise. In severe cases, a dysregulated “cytokine storm” contributes to acute respiratory distress syndrome (ARDS).
Antigenic Variation - Why Influenza Keeps Coming Back
Antigenic drift refers to the accumulation of point mutations in HA and NA genes over time. These gradual changes, concentrated in the antigenic sites that antibodies recognize, allow the virus to escape immunity generated by prior infection or vaccination. Because HA and NA are RNA polymerase products made by an error-prone enzyme (lacking proofreading), mutations occur constantly. This is why we need new flu vaccines each year - last year’s vaccine induces antibodies to last year’s HA and NA epitopes, which may no longer match this year’s circulating strains.
Antigenic shift is a sudden, major change in HA or NA, creating a novel subtype to which the population has no immunity. This occurs through reassortment - when two different influenza strains infect the same cell, their segmented genomes (8 separate RNA segments) can be packaged into progeny virions in new combinations. Pigs serve as “mixing vessels” because they can be infected by both avian and human strains, and their respiratory epithelium contains both α-2,3 and α-2,6-linked sialic acids. When a human virus and an avian virus co-infect a pig, reassortment can generate a virus with avian HA (to which humans have no immunity) but human-adapted internal genes (allowing efficient human-to-human transmission). The 1918 pandemic (H1N1), the 1957 pandemic (H2N2), and the 1968 pandemic (H3N2) all resulted from antigenic shift. The 2009 H1N1 “swine flu” pandemic arose through reassortment of avian, swine, and human influenza strains.
Types of Influenza Virus: Influenza A has 8 segments and infects multiple species (humans, birds, pigs, horses) - the only type that causes pandemics, because only influenza A has the animal reservoirs that allow antigenic shift. Classified by HA (H1-H18) and NA (N1-N11) subtypes. Influenza B has 8 segments but infects only humans/seals, undergoes only antigenic drift, and causes seasonal epidemics but not pandemics. Influenza C has 7 segments and causes mild respiratory illness (clinically minor).
Complications: the most common cause of death from influenza is post-influenza secondary bacterial pneumonia - from Staph aureus (including MRSA), Strep pneumoniae, H. influenzae, Strep pyogenes. Typical timeline: initial flu improves for 4-14 days, then recurrent fever and productive cough. Primary viral pneumonia, myocarditis, encephalitis, and Reye syndrome (with aspirin in children) are other complications.
Diagnostic specimens and methods:
- Nasopharyngeal swab is the preferred specimen
- PCR (RT-PCR) is the preferred method (most sensitive, can type/subtype)
- Rapid antigen (RIDT): fast but ~50-70% sensitive; negative should be confirmed by PCR in high-risk patients
- Hemadsorption - a historical culture-based test: RBCs are added to infected monolayers; if HA-expressing virus is present, RBCs stick to infected cells. Detects influenza, parainfluenza, and mumps (all HA-expressing). Largely replaced by PCR.
Treatment with neuraminidase inhibitors (oseltamivir, zanamivir, peramivir) works by blocking viral release. Baloxavir is a newer agent that inhibits the cap-dependent endonuclease required for viral mRNA synthesis. Treatment must be started within 48 hours of symptom onset. Amantadine/rimantadine (M2 channel blockers) are no longer used due to widespread resistance.
Influenza vaccines:
- Intramuscular inactivated (killed) - standard seasonal flu shot, ≥6 months, quadrivalent
- Intranasal live attenuated (LAIV/FluMist) - healthy non-pregnant ages 2-49; contraindicated in immunocompromised, pregnant, and young children
Coronaviruses
The Coronaviridae are enveloped, positive-sense ssRNA viruses with the largest RNA genome (~30 kb). Named for their crown-like spike proteins on EM. Human coronaviruses include seasonal common cold strains (229E, NL63, OC43, HKU1 - ~15-30% of colds, second to rhinovirus) and three epidemic/pandemic strains from bat reservoirs: SARS-CoV (2003), MERS-CoV (2012, intermediate host dromedary camels), SARS-CoV-2 (2019).
Structural proteins (SEMN):
- S (Spike): trimeric glycoprotein that mediates receptor binding and fusion. Primary target of vaccines and neutralizing antibodies.
- E (Envelope): small assembly/ion channel protein
- M (Membrane): most abundant, shapes the envelope
- N (Nucleocapsid): binds and packages the RNA genome
SARS-CoV-2 entry: spike protein binds ACE2 (angiotensin-converting enzyme 2), which is highly expressed on type II pneumocytes, enterocytes, endothelial cells, cardiac myocytes, and renal tubular cells. The host protease TMPRSS2 cleaves spike to enable membrane fusion. ACE2 is also the receptor for SARS-CoV (2003).
RNA replication: like all RNA viruses, coronaviruses use a virally encoded RNA-dependent RNA polymerase (RdRp), which host cells lack. This is the target of remdesivir (adenosine analog) and molnupiravir.
SARS-CoV-2 diagnostics:
- PCR (RT-qPCR) - gold standard, NP/OP swab
- Rapid antigen tests - detect N protein, sensitivity 50-80% vs PCR, best in symptomatic days 1-5. Negative antigen in a symptomatic patient should be confirmed by PCR.
- Antibody tests - limited acute utility; useful for seroprevalence and past infection. Anti-Spike (anti-S) = past infection OR vaccination (all major vaccines target spike). Anti-Nucleocapsid (anti-N) = natural infection only (N is not in vaccines). So anti-S+/anti-N- = vaccinated only; anti-S+/anti-N+ = prior infection.
Vaccines all target the spike protein:
- mRNA (Pfizer, Moderna) - lipid nanoparticle delivers spike mRNA. Does NOT enter nucleus or integrate into host DNA. Modified pseudouridine improves stability and reduces innate immune recognition.
- Viral vector (J&J, AstraZeneca) - adenovirus vector carrying spike gene
- Protein subunit (Novavax) - recombinant spike with adjuvant
HIV
HIV is a retrovirus (Retroviridae) - enveloped, positive-sense ssRNA, carries two copies of its RNA genome (diploid), and uses reverse transcriptase to create a DNA copy that integrates into the host genome. Understanding the molecular mechanisms of how HIV enters cells, how it destroys the immune system, and why it establishes an incurable latent reservoir is essential for understanding the disease and its treatment.
HIV belongs to the Lentivirus subfamily (“lenti” = slow). HIV-1 causes the global pandemic; HIV-2 is mostly West African, less pathogenic, slower progression, and less responsive to some antiretrovirals.
The Viral Structure and Key Proteins
HIV is an enveloped virus with a lipid membrane derived from the host cell. Embedded in this envelope are the spike proteins (gp120 and gp41) that mediate entry. Inside the virion is a cone-shaped capsid containing two copies of the RNA genome along with the enzymes reverse transcriptase, integrase, and protease - each a critical drug target.
Three main HIV genes:
- Gag (Group-specific Antigen) - encodes structural proteins: p17 (matrix), p24 (capsid - used in antigen testing), p7 (nucleocapsid)
- Pol (Polymerase) - encodes enzymes: reverse transcriptase, integrase, protease
- Env (Envelope) - encodes gp160 precursor, cleaved into gp120 (surface, binds CD4/coreceptor) and gp41 (transmembrane, fusion)
Plus 6 regulatory/accessory genes: tat, rev, nef, vif, vpr, vpu.
How HIV Enters Cells - The Molecular Dance of Infection
HIV entry is a multi-step process requiring interaction with two receptors, and understanding these steps explains both viral tropism and drug development.
Step 1: gp120 binds CD4. The viral envelope protein gp120 recognizes and binds to the CD4 receptor on the surface of T helper cells, macrophages, and dendritic cells. CD4 is normally used by these cells to interact with MHC class II molecules during antigen presentation - HIV essentially hijacks this immune recognition molecule. The gp120-CD4 binding triggers a conformational change in gp120 that exposes a previously hidden region called the V3 loop.
Step 2: Coreceptor binding. The conformationally changed gp120 now binds to a coreceptor - either CCR5 or CXCR4. These are chemokine receptors that normally guide immune cell migration. R5-tropic viruses (using CCR5) predominate during early infection and are the variants typically transmitted. X4-tropic viruses (using CXCR4) emerge later in infection and are associated with more rapid CD4 decline. Some viruses are dual-tropic. This coreceptor requirement explains why people homozygous for the CCR5-Δ32 deletion mutation are highly resistant to HIV infection - without CCR5, R5-tropic viruses cannot enter their cells. Maraviroc, a CCR5 antagonist, exploits this by blocking the coreceptor.
Step 3: gp41-mediated fusion. Coreceptor binding triggers another conformational change, now in gp41. This protein contains a fusion peptide that inserts into the host cell membrane. gp41 then folds back on itself, pulling the viral and cellular membranes together until they fuse, releasing the viral core into the cytoplasm. Enfuvirtide is a fusion inhibitor that blocks this step.
Why CD4 Cells Die - Multiple Mechanisms of Destruction
The progressive loss of CD4+ T cells is the hallmark of HIV disease, but the mechanisms are more complex than simple viral killing.
Direct viral cytopathic effect: When HIV replicates in a CD4+ T cell, viral budding from the cell membrane can be directly cytotoxic. High levels of intracellular viral proteins can trigger apoptosis. Accumulation of unintegrated viral DNA can activate DNA damage responses.
Immune-mediated killing: Many infected cells are killed by the host’s own immune response. CD8+ cytotoxic T lymphocytes recognize viral peptides presented on MHC class I molecules and kill infected cells. This is actually protective early in infection but contributes to CD4 loss over time. Antibody-dependent cellular cytotoxicity (ADCC) also destroys infected cells.
Bystander killing: The majority of CD4+ T cells that die during HIV infection may not be productively infected. Chronic immune activation leads to exhaustion and apoptosis of uninfected cells. Pyroptosis - an inflammatory form of cell death triggered by incomplete viral infection of resting CD4 cells - may be a major mechanism. When HIV enters a resting CD4 cell but cannot complete reverse transcription, the accumulated cytoplasmic DNA triggers inflammasome activation, caspase-1 activation, and pyroptosis, which releases inflammatory cytokines that activate and draw in more CD4 cells to be infected.
The Latent Reservoir - Why HIV Cannot Be Cured
Here is the central tragedy of HIV biology: the virus integrates its DNA into the host genome, and once integrated, it becomes part of the cell’s genetic material. In activated CD4 cells that are actively dividing, the virus replicates and the cell dies. But if an infected cell transitions to a resting memory state before being killed, the viral DNA goes dormant along with the cell’s transcription machinery. This creates a latent reservoir - long-lived memory CD4+ T cells harboring silent but replication-competent HIV provirus.
These latently infected cells persist for decades. They are invisible to the immune system because they don’t express viral proteins. They are invisible to antiretroviral drugs because they aren’t replicating. When the cell is reactivated years later, the virus can emerge. This reservoir is established within days of infection - even patients treated during acute infection retain a latent reservoir - and it is the fundamental barrier to cure.
Acute HIV (acute retroviral syndrome) presents 2-4 weeks after exposure as a flu-like/mononucleosis-like illness: fever, pharyngitis, lymphadenopathy, maculopapular rash, myalgia, oral ulcers, diarrhea. Viral load is extremely high but antibodies may not yet be detectable (window period). Distinguishing from EBV mono: oral ulcers are more prominent in acute HIV; Monospot is typically negative (can be false-positive). p24 antigen or HIV RNA detects acute HIV before antibodies develop.
The Diagnostic Algorithm
HIV diagnosis uses fourth- or fifth-generation testing as the first step.
- 4th generation HIV Ag/Ab ELISA detects both HIV-1/2 antibodies AND p24 antigen (capsid protein from gag). Combined result - you can’t tell which component is positive. Narrows window period to ~2 weeks. Sensitivity >99.9%.
- 5th generation differentiates p24 antigen from HIV-1 antibodies from HIV-2 antibodies in a single assay. This lets you recognize acute HIV (Ag+/Ab-) immediately.
If the screening test is positive, an HIV-1/HIV-2 antibody differentiation immunoassay follows. This confirms the result and distinguishes HIV-1 from HIV-2.
If the differentiation assay is negative or indeterminate but screening was positive, HIV-1 RNA testing (viral load) is performed. This detects acute HIV before antibody seroconversion.
Western blot is no longer the confirmatory test (historically required 2 of 3 bands: p24, gp41, gp120/160) - it has been replaced by the differentiation assay, which is faster and more sensitive for acute infection.
Neonates born to HIV+ mothers are not screened by ELISA because maternal IgG crosses the placenta and persists up to 18 months. Use proviral DNA PCR (detects integrated HIV DNA in infant cells, unaffected by maternal antibodies). Testing at birth, 14-21 days, 1-2 months, and 4-6 months. Two positive DNA PCRs confirm infection.
CD4 Count - The Measure of Immune Damage
HIV infects CD4+ T cells, and progressive CD4 depletion defines disease progression. In untreated infection, after an initial rapid decline during acute infection (when viral loads peak), the immune system partially contains the virus and CD4 counts plateau. This is followed by years of gradual decline at roughly 50-80 cells/μL per year. When the CD4 count falls below 200 cells/μL, the risk of opportunistic infections rises dramatically, and AIDS is defined.
HIV Opportunistic Infections by CD4 Threshold:
| <500 |
Kaposi sarcoma, TB reactivation |
- |
| <200 |
PCP (Pneumocystis), Candida esophagitis, Cryptococcus |
TMP-SMX |
| <150 |
Histoplasma, Coccidioides (disseminated) |
- |
| <100 |
Toxoplasma encephalitis, Cryptosporidium |
TMP-SMX (also covers Toxo) |
| <50 |
MAC (Mycobacterium avium complex), CMV retinitis, CNS lymphoma, PML |
Azithromycin (MAC) |
AIDS-defining illnesses (occurs at any CD4, but defines AIDS):
- PCP, Kaposi sarcoma, CMV disease, Cryptococcal meningitis
- Esophageal candidiasis, CNS lymphoma, Toxoplasma encephalitis
- Mycobacterial disease (TB, MAC), PML, Cryptosporidium/Isospora >1 month
- HIV encephalopathy, wasting syndrome, invasive cervical cancer
Viral Load - The Measure of Viral Activity
HIV-1 RNA (viral load) measures the amount of virus in blood and is measured by reverse transcriptase PCR. It’s used to diagnose acute infection (before antibodies develop), monitor treatment response, and assess adherence and resistance. The goal of treatment is undetectable viral load (<20-50 copies/mL, depending on assay). The “set point” viral load - the level several months after acute infection, once equilibrium is reached - predicts the rate of disease progression.
CD4+ T cell count is measured by flow cytometry using anti-CD3/CD4/CD8/CD45 antibodies. Normal CD4: 500-1500 cells/μL.
HIV Genotyping: identifies resistance mutations in RT, protease, and integrase genes by Sanger sequencing or NGS. Perform at time of diagnosis (baseline - 10-15% of new diagnoses have transmitted resistance) and at treatment failure (before changing regimen). Test while on the failing regimen, because resistant variants are replaced by wild-type once drug pressure is removed. HIV monotherapy and non-adherence drive resistance because incomplete suppression allows resistant mutants to emerge under selective pressure. This is why modern ART is 3-drug combination (typically 2 NRTIs + integrase inhibitor).
HIV Diagnosis in Neonates: Maternal IgG persists in the infant for up to 18 months → standard antibody tests (ELISA, Western blot) give FALSE POSITIVE results. Must use HIV NAT (DNA PCR or RNA viral load) tested at birth, 14-21 days, 1-2 months, and 4-6 months. Two positive NAT results confirm infection. Start zidovudine (AZT) prophylaxis within 6-12 hours of birth. No breastfeeding in resource-rich settings.
Antiretroviral Therapy - Attacking the Viral Life Cycle
Understanding how each drug class works requires understanding the viral replication steps:
Entry inhibitors block the steps described above: maraviroc blocks CCR5 coreceptor binding; enfuvirtide blocks gp41-mediated fusion.
NRTIs (nucleoside/nucleotide reverse transcriptase inhibitors) are nucleoside analogs that are incorporated into the growing DNA chain during reverse transcription and cause chain termination because they lack the 3’-hydroxyl group needed for the next phosphodiester bond.
NNRTIs (non-nucleoside reverse transcriptase inhibitors) bind directly to reverse transcriptase at a site away from the active site, causing conformational changes that inhibit enzyme function.
Integrase inhibitors block the integration of viral DNA into the host genome - a critical step for establishing both productive and latent infection. These are now first-line therapy due to excellent efficacy and tolerability.
Protease inhibitors block the viral protease that cleaves polyprotein precursors into functional proteins during viral maturation. Without this cleavage, virions bud but are non-infectious.
Combination therapy with drugs from multiple classes - typically two NRTIs plus an integrase inhibitor - achieves viral suppression, prevents resistance emergence, and has transformed HIV from a fatal illness to a manageable chronic condition.
HTLV-1 (Human T-lymphotropic Virus Type 1)
HTLV-1 is a deltaretrovirus (oncogenic retrovirus) that infects CD4+ T cells and causes disease after decades of latency. ~10-20 million infected worldwide. Endemic in:
- Japan (especially southwestern, Kyushu)
- Caribbean (Jamaica, Trinidad)
- Sub-Saharan Africa
- South America (Brazil), Middle East (Iran)
US blood banks screen donors for HTLV-1/2 antibodies.
Transmission (HTLV is cell-associated, so free virus is less infectious than HIV):
- Sex (male-to-female > female-to-male)
- Blood exposure - transfusion, IVDU. Leukoreduction partially reduces risk.
- Breastfeeding - the major vertical route in endemic areas (prolonged breastfeeding >6 months increases risk)
- Transplacental (less common than breastfeeding)
Primary infection is usually asymptomatic. Only 2-5% of carriers develop disease after 20-40 years of latency.
Oncogenesis: the Tax protein activates NF-kB, promotes T-cell proliferation, inhibits DNA repair, and inactivates p53. This drives polyclonal expansion of infected cells; additional genetic hits over decades produce a monoclonal malignancy.
Late sequelae:
- Adult T-cell leukemia/lymphoma (ATLL) - aggressive CD4+ T-cell malignancy. Latency 20-40 years. Lifetime risk ~5% if infected at <20 years old.
- Peripheral smear: flower (cloverleaf) lymphocytes - CD4+ T cells with deeply multilobated nuclei resembling flower petals
- Hypercalcemia (PTHrP from tumor cells, osteoclast activation) - “thirst”
- Elevated soluble IL-2 receptor (CD25)
- Skin lesions, lymphadenopathy, hepatosplenomegaly
- Despite elevated CD4 count, cells are dysfunctional (immunosuppression)
- IHC: CD4+, CD25+, CCR4+
- Acute subtype has median survival <1 year
- HTLV-1-associated myelopathy / Tropical spastic paraparesis (HAM/TSP) - immune-mediated demyelination of thoracic spinal cord (lateral columns, corticospinal tracts). Female:male ~3:1. Shorter incubation than ATLL, especially in transfusion-acquired cases (1-3 years). Progressive spastic paraparesis, urinary dysfunction.
- Strongyloides hyperinfection susceptibility - HTLV-1 impairs Th2 antihelminth defense. Screen HTLV-1 patients for Strongyloides and vice versa.
- Infectious dermatitis, uveitis
Hepatitis C Virus (HCV)
Hepatitis C is a flavivirus that has achieved something remarkable in modern medicine: it went from untreatable to curable within a few decades. The development of direct-acting antivirals (DAAs) transformed HCV from a leading cause of liver transplantation to an infection that can be cured in 8-12 weeks with oral therapy.
Transmission: primarily through infected blood - IV drug use (most common in developed countries), blood transfusion (historical, pre-1992 screening), needlestick, non-sterile tattooing/piercing, hemodialysis. Sexual transmission is rare (much less efficient than HBV/HIV). Vertical transmission ~5%. NOT transmitted by casual contact, food, water, or breastfeeding.
Natural history - these numbers are high-yield:
- ~75% develop chronic infection (contrast with HBV adult, where <10% become chronic)
- ~15% of chronic HCV patients develop cirrhosis over decades
- ~5% of chronic HCV patients develop hepatocellular carcinoma
Diagnosis uses a two-step approach:
- Anti-HCV antibody (ELISA/CLIA) - screening. Indicates exposure; persists even after the virus clears. Takes 4-10 weeks to appear after infection.
- If antibody positive, reflex to HCV RNA (quantitative PCR) to confirm active infection.
Interpretation: Ab+ / RNA+ = active infection. Ab+ / RNA- = resolved (spontaneous clearance or post-treatment SVR). In acute HCV or immunosuppressed patients, HCV RNA is the primary diagnostic test.
HCV RNA also establishes baseline viral load, monitors treatment response, confirms SVR, and detects relapse. ==Sustained virologic response (SVR) = undetectable HCV RNA at 12 or 24 weeks post-treatment == is the current definition of cure (concordance with long-term outcomes >99%).
HCV genotyping is performed at baseline. Seven genotypes; genotype 1 is most common worldwide and in the US (~70%). Subtype 1a is more likely to develop resistance (NS5A Q30R) than 1b. Pan-genotypic regimens (sofosbuvir/velpatasvir) have made genotyping less clinically essential.
Fibrosis staging: liver biopsy is not required. Non-invasive options: transient elastography (FibroScan) measures liver stiffness by ultrasound; FibroTest (serum markers); APRI; FIB-4. Cirrhotic patients still require HCC surveillance even after SVR.
Treatment has been revolutionized: DAAs target specific viral proteins (NS3/4A protease, NS5B polymerase, NS5A) and achieve SVR rates exceeding 95% with 8-12 weeks of oral therapy. The medications are well-tolerated and effective across all genotypes. The barrier to treatment is now primarily cost and access, not efficacy.
Hepatitis G (GBV-C / pegivirus) is another Flaviviridae transmitted parenterally. Found in 1-4% of blood donors; does NOT appear to cause hepatitis or liver disease. Interestingly, GBV-C coinfection in HIV patients is associated with slower HIV progression. No treatment indicated.
Other RNA Viruses
Rubella (German Measles) - Togaviridae
Togaviruses are enveloped, positive-sense ssRNA viruses with two clinically relevant genera: Alphavirus (arboviral encephalitides, chikungunya) and Rubivirus (rubella). Rubella is NOT arthropod-borne - it spreads by respiratory droplets.
Rubella must not be confused with rubeola (measles) - they’re completely different viruses causing different diseases, though both produce a rash. In children and adults, rubella is trivial. In fetuses, it’s devastating. This dichotomy explains why rubella vaccination is such a public health priority.
In children and adults, rubella causes mild illness: low-grade fever, malaise, and a fine maculopapular rash that starts on the face and spreads downward, lasting about 3 days (hence the old name “three-day measles”). The distinctive feature is lymphadenopathy, particularly involving posterior auricular, suboccipital, and posterior cervical nodes - finding these nodes in a patient with a viral exanthem should make you think of rubella. Arthralgias and arthritis are common in adult women. Patients are infectious from 7 days before to 7 days after rash.
Congenital rubella syndrome is the catastrophe: When rubella infects a pregnant woman, particularly in the first trimester, the consequences for the fetus are severe. Risk of congenital defects is ~85% with infection in first 12 weeks. Classic triad:
- Sensorineural deafness (most common, often the only manifestation, up to 80%)
- Cataracts (plus microphthalmos, glaucoma, “salt and pepper” retinopathy)
- Patent ductus arteriosus (plus peripheral pulmonary stenosis)
Other manifestations: microcephaly, intellectual disability, hepatosplenomegaly, thrombocytopenia with “blueberry muffin” rash, radiolucent bone disease. The tragedy is that this is entirely preventable through vaccination.
The MMR vaccine (measles, mumps, rubella) uses live attenuated virus and is contraindicated in pregnancy. All women of childbearing age should have rubella AND varicella IgG confirmed before pregnancy; vaccinate non-immune women at least 4 weeks before conceiving, or postpartum before discharge.
Alphaviruses - mosquito-borne:
- Eastern equine encephalitis (EEE) - most severe, mortality 30-50%, Culiseta mosquitoes, bird reservoir, Atlantic/Gulf coast
- Western (WEE) and Venezuelan (VEE) equine encephalitides - milder
- Chikungunya - Aedes aegypti and albopictus mosquitoes. High fever, maculopapular rash, and severe polyarthralgia that can persist months to years. Name means “that which bends up” (stooped posture). Same vectors as dengue and Zika.
Rotavirus - Reoviridae
Rotavirus was once the leading cause of severe childhood diarrhea worldwide, responsible for hundreds of thousands of deaths annually, primarily in developing countries. The introduction of rotavirus vaccines has dramatically reduced this burden - one of the great public health achievements of the 21st century.
Reoviruses are non-enveloped, segmented double-stranded RNA viruses (10-12 segments; “Reo” = Respiratory Enteric Orphan). The double-shelled icosahedral capsid gives rotavirus its wheel-like appearance on EM (hence “rota”). Being non-enveloped, rotavirus is environmentally stable and resistant to many disinfectants (ethanol-based sanitizers have limited efficacy).
Besides rotavirus, Reoviridae includes Coltivirus / Colorado tick fever virus - tick-borne (Dermacentor andersoni, western US/Canada mountain areas), flu-like illness with leukopenia and “saddleback” fever pattern. Differential includes Rocky Mountain spotted fever (same tick, same area, but Rickettsia rickettsii has rash and needs doxycycline).
The disease hits young children hardest: Rotavirus causes severe watery diarrhea and vomiting in children under 5, with peak incidence at 6-24 months. The illness lasts about a week, and the main danger is dehydration - in settings without access to rehydration therapy, this can be fatal. In temperate climates, rotavirus has a striking winter seasonality.
Vaccination has transformed rotavirus epidemiology: The oral rotavirus vaccines (Rotarix, RotaTeq) are live attenuated and given in infancy. Hospitalizations have declined by >80% in vaccinated populations. There is a small risk of intussusception, but the benefits outweigh this risk.
Diagnosis: PCR is most sensitive; stool antigen EIA is widely used in clinical practice. Included in most multiplex GI panels.
Astrovirus (Astroviridae): non-enveloped +ssRNA with a characteristic star-shaped appearance on EM (“astron”). Causes gastroenteritis primarily in infants <1 year old, elderly, and immunocompromised. Usually milder than rotavirus or norovirus. Diagnosis by stool PCR.
Hepatitis E (Hepeviridae): non-enveloped (quasi-enveloped in blood) +ssRNA. Transmitted fecal-oral (contaminated water, genotypes 1 and 2) or zoonotic from undercooked pork, deer, boar (genotypes 3 and 4, developed countries). Endemic in Southeast Asia, Africa, Central America, India.
- Usually self-limited acute hepatitis
- Characteristically severe in pregnant women, with mortality up to 20-25% for genotype 1 in the third trimester (fulminant hepatic failure with coagulopathy, encephalopathy)
- Can be chronic in immunosuppressed patients (genotype 3)
- Board pearl: pregnant woman with hepatitis and travel to Southeast Asia = hepatitis E
Norovirus - Caliciviridae
Norovirus (previously “Norwalk virus” after a 1968 Norwalk, Ohio outbreak) is the most common cause of acute viral gastroenteritis across all age groups and is notorious for causing explosive outbreaks in closed settings - cruise ships, nursing homes, hospitals, schools, and restaurants. Responsible for >50% of global gastroenteritis outbreaks. The virus combines several features that make it extraordinarily contagious.
Caliciviruses are non-enveloped, positive-sense ssRNA viruses named for the cup-shaped (calyx) surface depressions on EM.
What makes norovirus so transmissible: The infectious dose is tiny - as few as 18 viral particles can cause illness. The virus is shed in enormous quantities in stool and vomit. It’s non-enveloped, so it resists environmental inactivation and survives on surfaces for days. It can be transmitted by aerosolized vomitus. And people remain contagious for days after symptoms resolve.
The clinical picture is dramatic but brief: Onset is sudden - within 12-48 hours of exposure, patients develop projectile vomiting, watery diarrhea, abdominal cramps, and sometimes low-grade fever. The illness is miserable but self-limited, typically resolving in 24-72 hours. In healthy adults, it’s a bad couple of days. In the elderly or debilitated, dehydration can be dangerous.
Outbreak control is challenging: The combination of low infectious dose (~18 particles), environmental persistence (surfaces for weeks, survives freezing and 60C), and continued shedding makes norovirus outbreaks difficult to control. Hand hygiene must include soap and water - alcohol-based sanitizers are less effective against non-enveloped viruses. Environmental disinfection with bleach is necessary. In healthcare settings, cohorting patients and excluding symptomatic staff are essential.
Diagnosis: RT-qPCR of stool is the gold standard. Antigen tests have lower sensitivity (~50-75%). Norovirus cannot be grown in standard cell culture.
Flaviviruses - Mosquito-Borne Disease
The flaviviruses are a family of enveloped, positive-sense ssRNA viruses, most of which are arboviruses transmitted by mosquitoes or ticks. The name comes from yellow fever (flavus is Latin for yellow), the prototype of the family. HCV is also a Flaviviridae member but is blood-borne (not arboviral).
Important flaviviruses:
- Dengue - Aedes aegypti
- West Nile - Culex, bird reservoir
- Yellow fever - Aedes
- Zika - Aedes
- Japanese encephalitis - Culex, swine reservoir
- St. Louis encephalitis - Culex
- Tick-borne encephalitis, Powassan - ticks
- HCV (Hepacivirus) - blood-borne
Dengue fever affects hundreds of millions of people annually in tropical and subtropical regions. Aedes mosquitoes (the same genus that transmits Zika and chikungunya) are the vectors. Primary infection causes “breakbone fever” - high fever, severe headache, retro-orbital pain, and myalgias/arthralgias so severe they feel like bones breaking. Most patients recover completely.
The danger comes with secondary infection by a different dengue serotype (there are four). Antibodies from the first infection don’t neutralize the new serotype - instead, they enhance viral uptake into cells, a phenomenon called antibody-dependent enhancement. The result can be dengue hemorrhagic fever or dengue shock syndrome, with plasma leakage, hemorrhage, and potentially fatal shock.
Yellow fever remains endemic in sub-Saharan Africa and South America despite the existence of an excellent vaccine. Biphasic course: Phase 1 (infection, ~3-6 days) with fever, headache, chills, myalgia, N/V (most recover); Phase 2 (intoxication, ~15-25%) with high fever, jaundice, hepatic failure, hemorrhage/DIC, renal failure, and the classic “black vomit” (hematemesis). Phase 2 mortality 20-50%. Histology: Councilman bodies - eosinophilic apoptotic hepatocytes in mid-zonal hepatic necrosis. The live attenuated 17D vaccine provides lifelong immunity and is required for travel to endemic areas.
West Nile virus reached the United States in 1999 and has since spread across the country, transmitted by Culex mosquitoes, bird reservoir (corvids - crows, jays, magpies - are sentinel species; dead bird surveillance is used). Humans and horses are dead-end hosts. Most infections are asymptomatic. About 20% cause a flu-like illness. Less than 1% develop neuroinvasive disease - meningitis or encephalitis - but this can be severe, particularly in the elderly. Acute flaccid paralysis resembling polio can occur.
Zika virus gained global attention in 2015-2016 when the epidemic in Brazil revealed its ability to cause congenital Zika syndrome. In adults, infection is usually mild or asymptomatic - low-grade fever, rash, conjunctivitis, arthralgias. But when pregnant women are infected, the virus can cause devastating fetal brain damage, including microcephaly and other neurologic abnormalities. Zika can also be sexually transmitted, unlike most arboviruses.
Flavivirus diagnostics - an important topic because cross-reactivity makes serology tricky:
- PCR has a narrow window - viremia lasts only ~5-7 days after symptom onset. Use PCR in the first week; serology later. (Exception: Zika urine PCR may stay positive longer than blood.)
- MAC-ELISA (IgM antibody capture) - detects virus-specific IgM 3-8 days after symptoms. Main limitation: extensive cross-reactivity among flaviviruses (shared envelope epitopes). Prior dengue infection or yellow fever vaccination can cause false positives for Zika, West Nile, etc.
- Seroconversion or 4-fold IgM rise in paired sera 7-21 days apart is diagnostic.
- PRNT (plaque reduction neutralization test) is the gold-standard confirmatory test - measures virus-specific neutralizing antibodies by serial dilution incubated with live virus on cell monolayers. The virus with the highest neutralizing titer (lowest PRNT90 titer) is the infecting virus. Slow, BSL-3/4 required for some viruses.
- NS1 antigen rapid test is available for dengue.
Togaviruses - Alphavirus Encephalitides
Several alphaviruses cause encephalitis in the Americas, named for the equine hosts that amplify the viruses (horses, like humans, are dead-end hosts - they don’t transmit to mosquitoes efficiently).
Eastern equine encephalitis (EEE) is the most severe, with mortality rates around 30-50% and frequent neurologic sequelae in survivors. It’s found in the eastern United States and is fortunately rare because the mosquito vectors don’t commonly bite humans. Western equine encephalitis (WEE) and Venezuelan equine encephalitis (VEE) are less severe.
Chikungunya, also an alphavirus, has become globally important. It causes acute febrile illness with severe polyarthralgias - the joint pain is so characteristic that the name comes from a Tanzanian word meaning “to walk bent over.” Unlike dengue, chikungunya arthritis can persist for months to years in some patients.
Filoviruses - Ebola and Marburg
The filoviruses are among the most feared pathogens - their filamentous shape, high mortality, and dramatic hemorrhagic manifestations captured public attention during the 2014-2016 West African Ebola epidemic. Named for their filamentous (thread-like) morphology on EM - long pleomorphic filaments up to 14,000 nm in length. BSL-4 agents, CDC Category A bioterrorism. Reservoir: fruit bats.
Two genera: Ebolavirus (6 species, Zaire ebolavirus most lethal, up to 90% mortality) and Marburgvirus (first described 1967 in Marburg, Germany from imported African green monkeys).
Ebola and Marburg viruses are enveloped, negative-sense RNA viruses transmitted through contact with infected body fluids. In African outbreaks, transmission chains involve contact with sick family members, burial practices that involve touching corpses, and healthcare workers who lack adequate personal protective equipment. Not airborne transmitted.
Pathogenesis: the virus infects monocytes, macrophages, and dendritic cells, triggering massive cytokine release (“cytokine storm”) that causes endothelial dysfunction, vascular leak, and DIC.
The clinical course begins with nonspecific symptoms - fever, myalgia, headache - progressing to severe gastrointestinal symptoms with diarrhea and vomiting. Hemorrhagic manifestations (bleeding from multiple sites) occur late in the course and are not as universal as popular media suggests. Multi-organ failure and shock cause death; mortality varies by outbreak and viral strain but has ranged from 25% to 90%.
Treatment has improved dramatically. Supportive care - aggressive fluid resuscitation and electrolyte management - improves survival. Monoclonal antibody therapies (Inmazeb for Ebola Zaire) have shown mortality benefit and are now standard of care when available. The rVSV-ZEBOV (Ervebo) Ebola vaccine was approved in 2019.
Bunyaviruses - Hantavirus and Others
Bunyaviruses are enveloped, segmented (3 segments: L, M, S), negative-sense ssRNA viruses. Most are arthropod-borne; hantavirus is the notable exception (rodent-borne). Clinically important members: Hantavirus, Crimean-Congo hemorrhagic fever virus (tick-borne), Rift Valley fever virus (mosquito-borne), La Crosse encephalitis virus.
Hantaviruses are transmitted not by arthropods but by rodents. Humans become infected by inhaling aerosolized rodent urine, feces, or saliva - classically when cleaning out cabins or sheds with rodent infestations. There is no person-to-person transmission (rare exception: Andes virus). Different species carry different hantaviruses: deer mouse (Peromyscus maniculatus) carries Sin Nombre virus; striped field mouse carries Hantaan; bank vole carries Puumala.
In the Americas, hantaviruses cause hantavirus cardiopulmonary syndrome (HCPS) - Sin Nombre in the southwestern US (1993 Four Corners outbreak). After a prodromal illness with fever and myalgias, patients rapidly develop noncardiogenic pulmonary edema and cardiogenic shock from capillary leak. Mortality ~35%. ECMO may be life-saving.
Characteristic HCPS blood findings: neutrophilia without toxic granulation, thrombocytopenia, immunoblasts (>10% of lymphocytes), and hemoconcentration (elevated hematocrit from capillary leak). This triad of thrombocytopenia + immunoblasts + hemoconcentration in a patient with rapidly progressive pulmonary edema from a rural area is highly suggestive.
Old World hantaviruses (Hantaan, Seoul, Puumala) cause hemorrhagic fever with renal syndrome (HFRS) - fever, hemorrhage, acute kidney injury rather than pulmonary involvement.
California encephalitis virus (La Crosse virus) is a mosquito-borne bunyavirus causing encephalitis predominantly in children in the midwestern and eastern United States.
Arenaviruses - Rodent-Borne Viruses
Arenaviruses are enveloped, segmented ambisense RNA viruses (classified as negative-sense). Each segment (L and S) has both negative-sense and positive-sense coding regions. Named for the sandy granular appearance on EM (arena = sand in Latin) from incorporated host ribosomes. Two groups: Old World (LCMV, Lassa) and New World (Junin, Machupo, Guanarito - South American hemorrhagic fevers). Rodent reservoirs with chronic, asymptomatic infection.
Lymphocytic choriomeningitis virus (LCMV) is carried by the common house mouse (Mus musculus) and hamsters, transmitted through aerosolized excreta. Causes a biphasic illness: Phase 1 flu-like (fever, malaise, myalgia), then Phase 2 aseptic meningitis. CSF can show markedly low glucose - unusual for viral meningitis and can mimic TB meningitis. Most cases resolve. LCMV infection during pregnancy can cause congenital infection with severe neurologic damage, mimicking TORCH. In transplant recipients (organs from infected donors), LCMV causes fatal disease - a recognized cluster entity.
Lassa fever is a severe hemorrhagic fever caused by Lassa virus, endemic to West Africa (Sierra Leone, Liberia, Nigeria, Guinea). Reservoir: Mastomys rat (contact with urine/feces). Gradual onset fever, malaise, pharyngitis progressing to facial/neck edema, pleural effusion, hemorrhage. Mortality ~1% overall, 15-20% hospitalized. Deafness is a common sequela (~30%). Treatment: ribavirin (most effective when given early). Person-to-person transmission occurs. BSL-4.
Poxviruses - The Largest and Most Complex Viruses
Poxviruses are extraordinary in virology - they’re the largest viruses known, visible under light microscopy, and they possess a complexity that approaches that of small bacteria. They’re large enough to carry genes for their own DNA replication machinery, which is why they uniquely replicate entirely in the cytoplasm rather than requiring access to the nucleus like all other DNA viruses. This cytoplasmic replication creates the characteristic cytoplasmic inclusions seen on histopathology.
Smallpox (Variola Virus)
Smallpox holds a unique place in medical history as the only human infectious disease to be deliberately eradicated through vaccination - a campaign completed in 1980. The disease was devastating: Variola major mortality 30%; Variola minor (alastrim) ~1%. Survivors were often disfigured. The last natural case was in 1977 in Somalia. The virus now exists only in two WHO-authorized repositories: CDC (Atlanta) and VECTOR (Novosibirsk, Russia). It remains a CDC Category A bioterrorism agent.
CDC Category A bioterrorism agents (highest priority):
- Variola (smallpox)
- Bacillus anthracis (anthrax)
- Clostridium botulinum toxin (botulism)
- Francisella tularensis (tularemia)
- Yersinia pestis (plague)
- Filoviruses (Ebola, Marburg) - hemorrhagic fever
- Arenaviruses (Lassa, Machupo) - hemorrhagic fever
Pathogenesis: Variola virus was transmitted by respiratory droplets and aerosols. After inhalation, the virus replicated in the upper respiratory tract, then spread to regional lymph nodes. Primary viremia seeded the reticuloendothelial system (spleen, liver, bone marrow), where massive replication occurred. Secondary viremia then disseminated the virus to the skin and mucous membranes, producing the characteristic rash.
The rash was distinctive: lesions evolved synchronously through stages (macule → papule → vesicle → pustule → crust), so at any given time, all lesions were at the same stage. This synchronous evolution distinguishes smallpox from chickenpox, where lesions appear in successive crops and exist in multiple stages simultaneously. The centrifugal distribution - denser on face and extremities than trunk, including palms and soles - was also characteristic. The rash classically starts as an enanthem in the oral cavity before skin lesions appear. Smallpox lesions are deep and firm; chickenpox lesions are superficial and thin-walled.
| Lesion stages |
All same age (synchronous) |
Different ages (crops) |
| Distribution |
Centrifugal (face/extremities > trunk) |
Centripetal (trunk > extremities) |
| Palms/soles |
Involved |
Spared |
| Depth |
Deep, firm |
Superficial, thin-walled |
Guarnieri bodies are the pink eosinophilic cytoplasmic inclusions seen in poxvirus-infected cells - aggregates of viral replication machinery (viral factories) within the cytoplasm. Unique among DNA viruses, poxviruses replicate entirely in the cytoplasm (they carry their own DNA-dependent RNA polymerase). EM shows characteristic brick-shaped/ovoid particles with a dumbbell-shaped core.
Vaccinia is a closely related orthopoxvirus that produces mild disease and is the smallpox vaccine, cross-protective against variola and monkeypox. Administered by scarification with a bifurcated needle; a successful “take” produces a pustule that scars. Complications include progressive vaccinia (in immunocompromised), eczema vaccinatum, post-vaccinial encephalitis, and myocarditis.
Bioterrorism concern: Smallpox is classified as a Category A bioterrorism agent because of its transmissibility, high mortality in an unvaccinated population, and potential for public panic. Routine vaccination ended in the 1970s, leaving most of the world’s population susceptible. The Strategic National Stockpile maintains enough vaccine for emergency response.
Molluscum Contagiosum
Molluscum contagiosum virus causes a benign skin infection that contrasts starkly with the severity of smallpox - a reminder of how diverse poxvirus pathogenicity can be.
Pathogenesis: Molluscum spreads through direct skin contact and causes highly localized infection of the epidermis. The virus infects keratinocytes and induces them to proliferate, creating the characteristic lesion: a dome-shaped, waxy papule with a central umbilication (dimple). This umbilicated center contains the viral-laden central core. The virus produces proteins that interfere with interferon signaling and antigen presentation, allowing it to evade local immunity and persist for months.
Clinical patterns: In immunocompetent children, molluscum is a common, self-limited infection acquired by skin contact. Lesions resolve spontaneously over 6-12 months as cellular immunity eventually clears the infection. In adults, molluscum is often sexually transmitted, with genital distribution. In AIDS patients with advanced immunosuppression, molluscum can be extensive and disfiguring, with hundreds of large lesions - this extensive pattern can be an AIDS-presenting illness.
Henderson-Patterson bodies (also called molluscum bodies) are the diagnostic histopathologic finding: large, eosinophilic, intracytoplasmic inclusions that push the nucleus to the periphery of the cell. They represent viral inclusion bodies filled with maturing virions. Histologically, the lesion is a cup-shaped/crateriform epithelial proliferation with central umbilication, and the molluscum bodies enlarge progressively from basal to superficial layers (largest in the granular/cornified layers, where they are extruded through the umbilication). Keratohyalin granules are prominent and coarse.
Molluscum in different hosts:
- Children (1-10 years): trunk/extremities/face, direct contact or fomites, self-limited in 6-18 months.
- Adults: consider molluscum an STI when genital/inguinal. Test for other STIs and HIV.
- HIV/AIDS with CD4 <200: giant molluscum (>1 cm), widespread, resistant to treatment, often on face. Differential for multiple facial umbilicated papules in HIV: molluscum, disseminated cryptococcosis, disseminated histoplasmosis, Penicillium (Talaromyces) marneffei. Giant facial molluscum in an adult should prompt HIV testing.
Treatment is not mandatory since lesions self-resolve, but can include curettage, cryotherapy, or topical agents to hasten clearance.