My PhD thesis applied statistical genetics methods to understand lung cancer. This page walks through the field from first principles: what genetic data looks like, how we find disease associations, why those associations are tricky to interpret, and how we can use genetics to ask causal questions.
Part 1: The Basics
What is DNA?
DNA is a molecule made of four chemical bases: Adenine, Thymine, Guanine, and Cytosine. Your genome is a sequence of about 3 billion of these letters, split across 23 pairs of chromosomes.
The key fact: 99.9% of your DNA is identical to everyone else’s. The interesting part is the 0.1% where we differ.
What is a SNP?
A SNP (single nucleotide polymorphism, pronounced “snip”) is a position in the genome where people have different letters. At most positions, everyone has the same base. But at about 4-5 million positions, there’s variation.
html`<div style="font-family: system-ui, sans-serif; max-width: 700px; margin: 20px 0; padding: 20px; background: #fefce8; border-radius: 8px;"> <svg viewBox="0 0 650 200" style="width: 100%; height: auto;"> <text x="20" y="25" font-size="14" fill="#854d0e" font-weight="bold">A SNP is a position where people differ:</text> <!-- Person 1 --> <text x="20" y="65" font-size="13" fill="#666">Person 1:</text>${["A","T","G","C","C","A","T","G","G","A","C","T"].map((base, i) => {const colors = {A:"#ef4444",T:"#22c55e",G:"#3b82f6",C:"#f59e0b"};const isSNP = i ===5;returnsvg` <rect x="${100+ i *28}" y="${45}" width="25" height="28" rx="3" fill="${isSNP ?'#fef08a':'#f8fafc'}" stroke="${isSNP ?'#ca8a04':'#e2e8f0'}" stroke-width="${isSNP ?2:1}"/> <text x="${112+ i *28}" y="66" text-anchor="middle" font-size="15" fill="${colors[base]}" font-weight="bold" font-family="monospace">${base}</text> `; })} <!-- Person 2 --> <text x="20" y="115" font-size="13" fill="#666">Person 2:</text>${["A","T","G","C","C","G","T","G","G","A","C","T"].map((base, i) => {const colors = {A:"#ef4444",T:"#22c55e",G:"#3b82f6",C:"#f59e0b"};const isSNP = i ===5;returnsvg` <rect x="${100+ i *28}" y="${95}" width="25" height="28" rx="3" fill="${isSNP ?'#fef08a':'#f8fafc'}" stroke="${isSNP ?'#ca8a04':'#e2e8f0'}" stroke-width="${isSNP ?2:1}"/> <text x="${112+ i *28}" y="116" text-anchor="middle" font-size="15" fill="${colors[base]}" font-weight="bold" font-family="monospace">${base}</text> `; })} <!-- Arrow pointing to SNP --> <line x1="240" y1="135" x2="240" y2="155" stroke="#ca8a04" stroke-width="2"/> <polygon points="235,155 245,155 240,165" fill="#ca8a04"/> <text x="240" y="185" text-anchor="middle" font-size="13" fill="#854d0e" font-weight="bold"> This is a SNP </text> <text x="240" y="200" text-anchor="middle" font-size="11" fill="#666"> Person 1 has "A", Person 2 has "G" </text> </svg></div>`
At this SNP, some people have an A and others have a G. We call these the two alleles (versions) of the SNP.
How do we measure SNPs? Genotyping arrays
We can’t afford to sequence everyone’s full genome (3 billion letters). Instead, we use genotyping arrays (also called SNP chips or microarrays) that measure only the positions where people differ - typically 500,000 to 2 million SNPs.
The output is a big matrix: rows are SNPs, columns are people, values are 0/1/2. With modern arrays and biobanks, we have data on millions of SNPs across hundreds of thousands of people.
Part 2: Finding Disease Variants
The question: which SNPs affect disease?
Now we have genetic measurements. The natural question: are certain variants more common in people with a disease?
This is what a genome-wide association study (GWAS) answers.
How GWAS works
For each SNP, we compare the frequency of each allele in cases (people with disease) versus controls (people without disease). If one allele is significantly more common in cases, that SNP is “associated” with the disease.
We do this test for every SNP - typically 500,000 to 5 million tests. Because we’re doing so many tests, we need a very stringent significance threshold (p < 5×10⁻⁸) to avoid false positives.
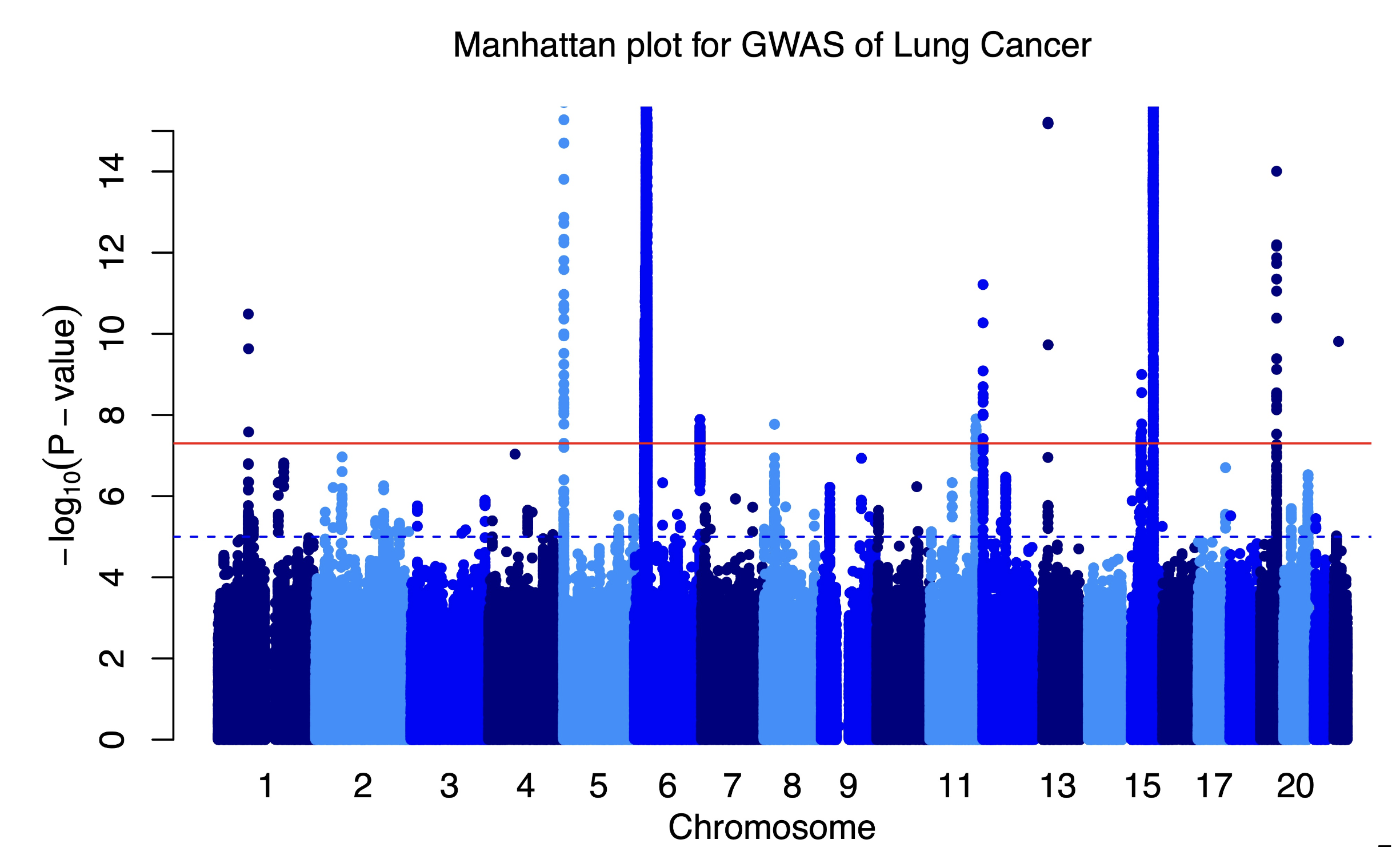
Manhattan plots: visualizing GWAS results
The results are displayed in a “Manhattan plot” - named because the peaks look like a city skyline.
Manhattan plot showing GWAS results
Each dot is one SNP. The x-axis shows position in the genome (chromosomes 1-22 plus X). The y-axis shows how strongly that SNP is associated with the disease (-log10 of the p-value, so higher = more significant).
Peaks above the red significance line are “hits” - SNPs associated with the disease.
Part 3: The LD Problem
So far so good - run a GWAS, find associated SNPs, done. But here’s the complication: those peaks in the Manhattan plot aren’t single SNPs. They’re clusters. Why?
Chromosomes are inherited in chunks
Each parent has two copies of each chromosome. When making eggs or sperm, these two copies can swap pieces - this is called recombination. The child then gets one (possibly recombined) chromosome from each parent.
But recombination is rare - only about 1-2 crossovers per chromosome per generation. So nearby regions of the chromosome travel together.
What this creates in a population: linkage disequilibrium
Over many generations, this creates a pattern called linkage disequilibrium (LD): nearby SNPs are correlated. If you know someone’s genotype at one SNP, you can often predict their genotype at nearby SNPs.
If one SNP actually causes disease, all the nearby SNPs that are correlated with it will also appear associated with the disease - even though they’re just bystanders.
gwasSignals2 = [ {pos:-100,signal:2,isCausal:false},// far - gray {pos:-60,signal:4.5,isCausal:false},// medium - orange {pos:-30,signal:7,isCausal:false},// close - orange {pos:0,signal:10,isCausal:true},// causal - red {pos:30,signal:7,isCausal:false},// close - orange {pos:60,signal:4.5,isCausal:false},// medium - orange {pos:100,signal:2,isCausal:false},// far - gray]html`<div style="font-family: system-ui, sans-serif; max-width: 700px; margin: 20px 0; padding: 20px; background: #fef2f2; border-radius: 8px;"> <p style="margin: 0 0 10px 0; font-size: 14px; color: #991b1b; font-weight: bold;">One causal SNP creates many "hits"</p> <svg viewBox="0 0 600 240" style="width: 100%; height: auto;"> <!-- Significance line at signal = 3 --> <line x1="50" y1="130" x2="550" y2="130" stroke="#dc2626" stroke-width="2" stroke-dasharray="6,4"/> <text x="555" y="134" font-size="10" fill="#dc2626">significance threshold</text> <!-- Axes --> <line x1="50" y1="180" x2="550" y2="180" stroke="#666" stroke-width="1.5"/> <line x1="50" y1="180" x2="50" y2="30" stroke="#666" stroke-width="1.5"/> <text x="300" y="200" text-anchor="middle" font-size="11" fill="#666">Position on chromosome</text> <text x="25" y="105" font-size="11" fill="#666" transform="rotate(-90, 25, 105)">-log(p-value)</text> <!-- SNP dots - signal * 15 for y position, threshold at signal=3 means y=135 -->${gwasSignals2.map(snp =>svg` <circle cx="${300+ snp.pos*2}" cy="${180- snp.signal*15}" r="${snp.isCausal?14:10}" fill="${snp.isCausal?'#dc2626': snp.signal>3?'#fb923c':'#94a3b8'}"/> `)} <!-- Legend --> <rect x="50" y="210" width="500" height="26" rx="4" fill="#fef2f2"/> <circle cx="90" cy="223" r="7" fill="#dc2626"/> <text x="105" y="227" font-size="11" fill="#333">Causal SNP</text> <circle cx="230" cy="223" r="7" fill="#fb923c"/> <text x="245" y="227" font-size="11" fill="#333">Correlated SNPs (in LD) - also significant!</text> <circle cx="470" cy="223" r="7" fill="#94a3b8"/> <text x="485" y="227" font-size="11" fill="#333">Far away</text> </svg></div>`
This is why Manhattan plots have broad peaks, not single spikes. Each peak is one (or a few) causal variants plus many correlated neighbors that get “dragged along” due to LD.
Quantifying LD
We measure LD with a statistic called r²:
r² = 1: Two SNPs are perfectly correlated (always inherited together)
r² = 0: Two SNPs are independent (no correlation)
LD decays with distance - nearby SNPs have high r², distant SNPs have low r².
So we’ve run a GWAS and have thousands of associated SNPs. But many are just correlated with causal variants (the LD problem), and we want to understand the bigger picture. What can we actually learn?
LD Score Regression (LDSC)
LDSC is a method that turns the LD problem into a feature. Instead of being confused by correlated SNPs, we use the pattern of correlations to learn about the trait.
The key insight: If a trait is truly affected by many variants across the genome (polygenic), then SNPs in high-LD regions should show stronger GWAS signals on average. Why? Because a SNP in a high-LD region is correlated with more of its neighbors, so it “tags” more potential causal variants.
Slope = heritability signal: If a trait is affected by many genetic variants, SNPs in high-LD regions will tag more of them, so their χ² statistics will be higher. The steeper the slope, the more heritable the trait.
Intercept = confounding check: The intercept should be ~1 if there’s no confounding. If it’s elevated, something other than genetics (like population structure or cryptic relatedness) is inflating results.
But the real power is comparing traits. If we have GWAS results for two different traits, we can ask: do the same SNPs tend to affect both?
This is powerful: we can measure the genetic overlap between any two traits that have been studied with GWAS - even if they were measured in completely different people.
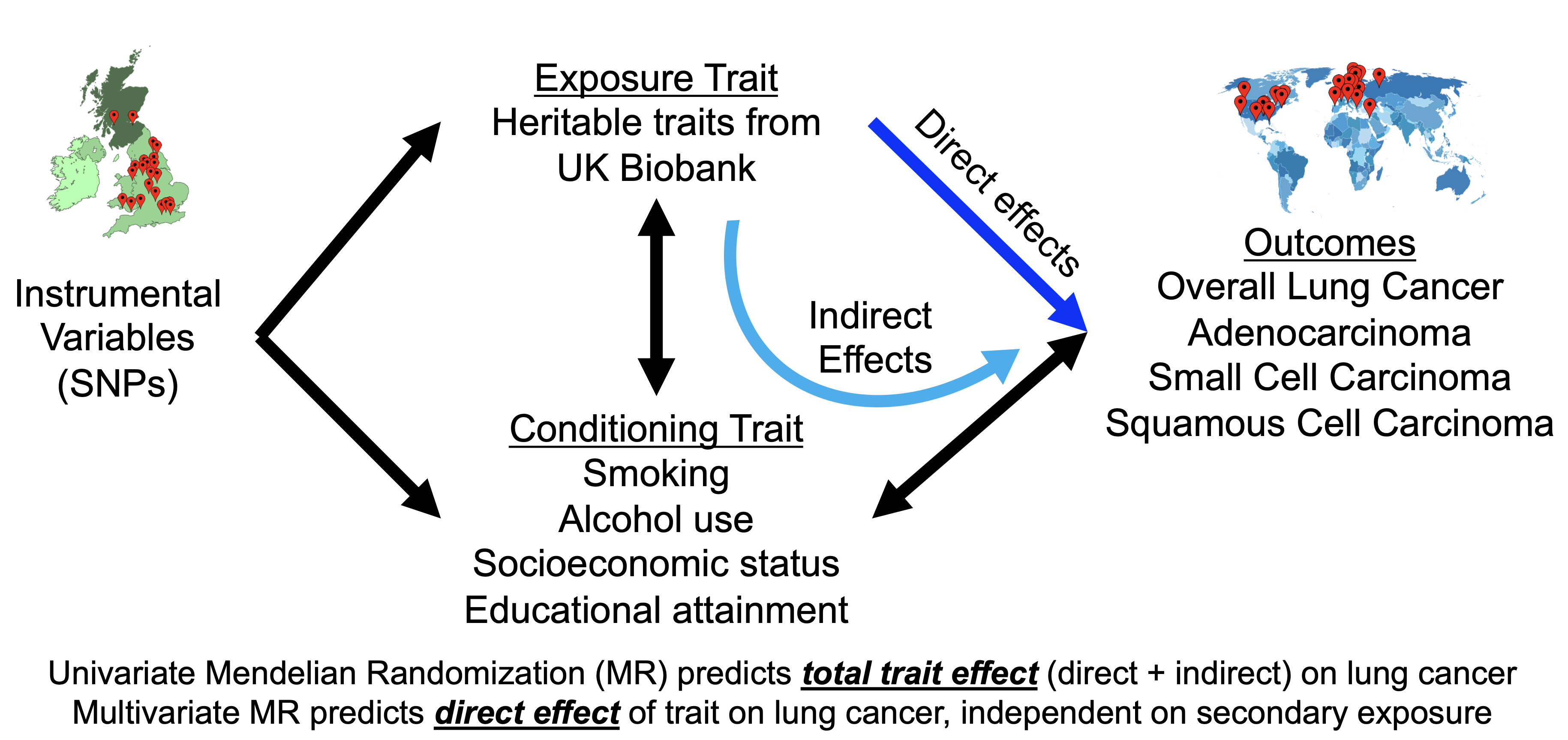
Mendelian Randomization: From Correlation to Causation
GWAS and LDSC tell us about associations. But the question we really want to answer is: does one thing cause another?
Observational studies struggle with this. If smokers have higher lung cancer rates, is it because smoking causes cancer? Or because the same factors (stress, socioeconomic status, personality) lead to both smoking and cancer?
Mendelian randomization (MR) uses genetics to cut through this problem.
The core insight: Your genetic variants were assigned randomly at conception - like a natural coin flip. Unlike your behaviors or environment, your DNA wasn’t shaped by confounding factors. This makes genetics a powerful tool for causal inference.
The three assumptions (all must hold for valid inference):
Relevance: The genetic variants must actually affect the exposure (smoking). We verify this with strong GWAS associations.
Independence: The variants can’t be associated with confounders. Because genotypes are assigned at conception before any environmental exposures, this usually holds.
Exclusion restriction: The variants can only affect the outcome (cancer) through the exposure (smoking) - not through other pathways. This is the trickiest assumption. A SNP that affects both smoking and lung function directly would violate it.
We use GWAS “summary statistics” - publicly available results showing how each SNP associates with a trait. We don’t need individual-level data. For MR, we need:
GWAS results for the exposure (e.g., smoking behavior)
GWAS results for the outcome (e.g., lung cancer)
For each SNP that affects smoking, we ask: does its effect on smoking predict its effect on cancer?
Mendelian randomization analysis
Reading an MR plot: Each point is a SNP. The x-axis shows how much that SNP affects the exposure (smoking), and the y-axis shows how much it affects the outcome (lung cancer). If smoking causes cancer, the points should fall along a line - SNPs that increase smoking more should proportionally increase cancer more. The slope of that line estimates the causal effect.
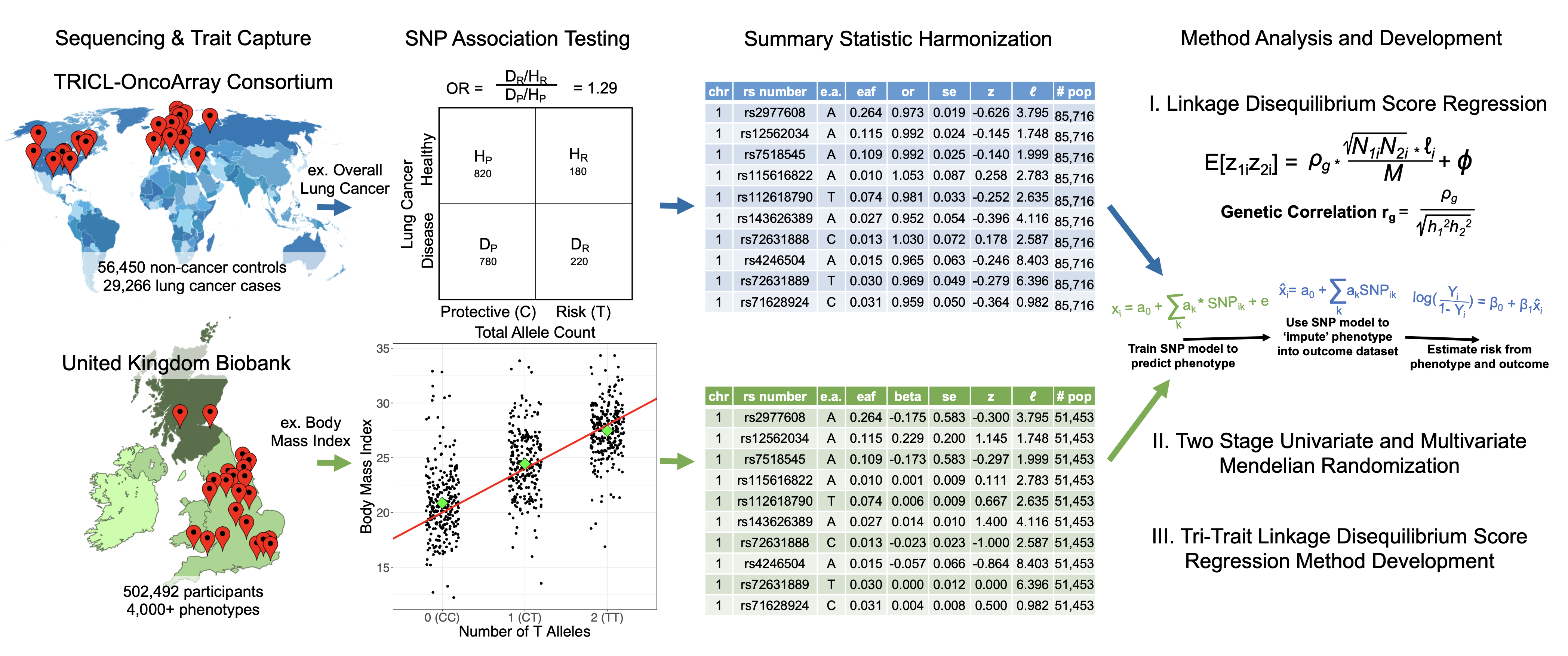
I applied these methods to lung cancer genetics using data from:
TRICL-OncoArray: ~30,000 lung cancer cases, 56,000 controls
UK Biobank: 500,000 people with extensive phenotype data
What I found
Genetic architecture varies by subtype: Lung adenocarcinoma, squamous cell carcinoma, and small cell carcinoma have different genetic profiles and different degrees of smoking-mediated effects.
Direct vs. mediated effects: Some genetic variants affect lung cancer risk through smoking behavior. Others affect it directly. I developed methods to distinguish these.
Causal relationships: Using MR, I quantified causal effects of smoking, education, and alcohol on lung cancer risk across subtypes.
I also developed a tri-trait method for computing genetic correlations while accounting for confounding by a third trait.
Publications
Pettit, R., Byun, J., Han, Y., et al. (2023). “Heritable traits and lung cancer risk: a two-sample mendelian randomization study.” Cancer Epidemiology, Biomarkers & Prevention, 32(10), 1421-1435.
Pettit, R., & Amos, CI. (2022). “Linkage disequilibrium score statistic regression for identifying novel trait associations.” Current Epidemiology Reports, 9(3), 190-199.
Pettit, R., Byun, J., Han, Y., et al. (2021). “The shared genetic architecture between epidemiological and behavioral traits with lung cancer.” Scientific Reports, 11(1), 17559.
Mentorship
I worked with Prof. Chris Amos at Baylor College of Medicine (profile).